
Abstract
Cellular senescence is a key physiological barrier against tumor and represents an option for therapeutic intervention. One pivotal intracellular stimulus causing senescence is DNA damage response, while the senescence-associated heterochromatin in cancer limits the strength of the DNA damage response to endogenous genotoxic stress or DNA-damaging agents. Therefore, targeting the maintenance of compacted chromatin in cancer cells represents an optional intervention to improve the therapeutic efficacy in cancer treatment. Given a crosstalk between methionine cycle and histone methylation, we hypothesize that pharmacologically disrupting methylation potential, defined as the ratio of cellular S-adenosylmethionine to S-adenosylhomocysteine, could affect the chromatin structures in cancer cells and thus enhance their sensitivity to DNA damage response signaling. Our results showed that 3-deazaneplanocin A, a chemical inhibitor of S-adenosylhomocysteine hydrolase, elicited a typical cellular senescence in hepatoma cells. Therapy-induced senescence by 3-deazaneplanocin A was mediated through p53–p21 pathway and triggered by enhanced ataxia-telangiectasia mutated activation related to chromatin changes. In conclusion, our study demonstrated that metabolic perturbation of chromatin status in oncogene-activated cancers could be an optional intervention to sensitize DNA damage response signaling.
Introduction
An emerging anticancer approach is the induction of cellular senescence, which was originally described as proliferative arrest due to the erosion of telomeres after a limited number of cell divisions in primary normal cells. 1 Similar to the natural replicative senescence, a comparable premature or accelerated senescence has been identified as a component of the tumor cell response to chemotherapeutic drugs or radiation. 2 Accumulating evidence indicates that senescence-inducing agents provide equivalent or prolonged survival with less adverse effects compared with other cytotoxic agents. 3 However, one factor limiting this strategy of cancer therapy is the lack of efficient senescence-inducing agents in clinical practice.
One pivotal intracellular stimulus causing senescence is DNA damage response (DDR), 4 which is substantially affected by the status of chromatin in cancer cells. 5 DDR is a cellular signaling cascade that senses and responds to genotoxic stress and includes collective events which lead to cell cycle arrest or DNA repair. At the initial phase of oncogene activation, cells under replicative stress trigger the activation of DDR signaling and establish the senescence associated with increased heterochromatin to form a barrier to transformation into cancer cells. 6 However, once the cells escape senescence and fully transform, those established senescence-associated heterochromatin foci (SAHF) will facilitate cancer cells to decrease their sensitivity to DNA-damaging stimuli. 7 Therefore, interventions to perturb the maintenance of SAHF in cancer cells might enhance both endogenous and therapy-induced DDR signaling to result in cellular senescence or apoptosis.
The maintenance of chromatin status relies on various post-translational modifications of histones. In particular, histone methylation such as the trimethylated form of Lys 9 on histone H3 (H3K9me3) directly involves in the formation of global heterochromatin or SAHF in cancer cells. 8 Histone methyltransferases (HMTs) utilize S-adenosylmethionine (SAM) to donate a methyl group to their histone substrates, and the produced S-adenosylhomocysteine (SAH) in turn acts as a negative feedback regulator for HMTs. 9 By modulating the balance between SAM and SAH levels, the status of methionine metabolism at diet level is sufficient to determine levels of histone methylation. 10 Therefore, we hypothesize that pharmacologic perturbation of methionine cycle would preferentially affect the chromatin structures of those cancer cells deeply dependent on SAHF and subsequently enhance their sensitivity to endogenous DDR signaling or DNA-damaging agents. Here, we report that pharmacologic inhibition of S-adenosylhomocysteine hydrolase (SAHH) by its potent inhibitor 3-deazaneplanocin A (DZNep) induces senescence in tumor cells and thus serves as a promising DDR-enhancing drug.
Materials and methods
Cells and drug treatments
HepG2 cells were purchased from the Shanghai Institute of Cell Biology (Shanghai, China) and cultured in Dulbecco’s Modified Eagle Medium containing 5% fetal bovine serum (FBS) and incubated at 37°C in a humidified atmosphere containing 5% CO2. Cells were seeded at 5000/cm2 the day before the drugs were added. The drug DZNep (Selleckchem, Houston, TX, USA) was dissolved in dimethyl sulfoxide (DMSO) and GSK343 (Selleckchem) in alcohol. Doxorubicin (Dox; Sigma–Aldrich, St Louis, MO, USA) was dissolved in distilled water.
Cell proliferation assays
Cells at the density of 5000/cm2 on a six-well plate were treated with drugs at a serial of concentrations for 5 days. Cell viability was assayed by the CellTiter 96 Aqueous One Solution Assay (MTS) kit (Promega, Madison, WI, USA) following the instructive manual.
Detection of SAH accumulation in cells
The details can be seen in Supporting Information Doc. S1.
Senescence-associated β-galactosidase assays
Cellular senescence was assayed using Senescence β-Galactosidase Staining Kit (Cell Signaling, Danvers, MA, USA) following the manufacturer’s instructions.
Cell cycle assay
For cell cycle assay, cells were harvested by trypsin at the indicated time points, washed in ice-cold phosphate-buffered saline (PBS), and fixed in 1 mL of ice-cold 70% ethanol overnight. After removing ethanol, the cells were stained in the dark with a solution containing 50 µg/mL propidium iodide (PI; Sigma–Aldrich) and 100 µg/mL RNase for 30 min at room temperature. The stained cells were analyzed for DNA content by BD FACSCanto II flow cytometry (BD Biosciences, San Diego, CA, USA).
RNA isolation and quantitative real-time polymerase chain reaction
The expression profiles of genes were analyzed by quantitative real-time polymerase chain reaction (RT-PCR) analysis. Total RNA was isolated using TRIzol (Life Technologies, Carlsbad, CA, USA) from HepG2 cells. Then RNA was reverse transcribed to complementary DNA (cDNA) using HiFiScript 1st Strand cDNA Synthesis Kit (CW Biotech, Beijing, China). The SYBR-green method was used for the relative quantification of messenger RNA (mRNA) expression. The primer sequences were retrieved for PrimerBank database according to the official gene names. 11 The values were normalized to the levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA and were analyzed by comparative CT method. PCRs were performed on Applied Biosystems ViiA System (Life Technologies).
Western blots
Cells were rinsed with PBS and scraped into radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with protease inhibitors (CW Biotech). Equal amounts (20–50 µg) of protein were separated on sodium dodecyl sulfate (SDS)–polyacrylamide gels and transferred to polyvinylidene difluoride membranes. Proteins were immunodetected using specific antibodies against β-actin (1:3000; CW Biotech; 0096), p16 (1:2000; Abcam; ab32034; Cambridge, MA, USA), p21 (1:100; Abclonal; AP0327; Wuhan, China), p27 (1:1000; Abcam; ab108349), p53 (1:50; Thermo Fisher Scientific; RM-9105; Fremont, CA, USA), EZH2 (1:500; Active Motif; 39902; Carlsbad, CA, USA), and DNA damage antibody including phospho-ataxia-telangiectasia mutated (ATM) (Ser1981), H2AX (Ser139), Chk1 (Ser345), and Chk2 (Thr68) (1:1000; Cell Signaling; 9947) overnight at 4°C. After washing with Tris buffered saline with Tween 20 (TBST) buffer, the membranes were incubated with appropriate secondary antibodies for 1 h at room temperature. Immunoblots were detected using an eECL Western Blot Kit (CW Biotech) following the manufacturer’s instruction.
Immunofluorescence cell staining
Cells were fixed in 4.0% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked in 1.0% bovine serum album in PBS for 30 min at room temperature. Next, the cells were incubated at 4°C overnight with the primary antibodies for p53 (1:100; Thermo Fisher Scientific; RM-9105). Alexa Fluor 546 Donkey Anti-Rabbit IgG (Thermo Fisher Scientific; A10040) was applied to the cells for 1 h at room temperature. Cell nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI; Roche, Nutley, NJ, USA). The cells were analyzed with a fluorescent microscope (Olympus, IX71, Japan).
Microarray analysis
HepG2 cells were treated with 5.0 µmol/L DZNep and 15 µmol/L GSK343 for 72 h. Untreated cells were used as the control. Whole-genome expressions were analyzed on microarray (Roche-Nimblegen; 05543789001) by KangChen Bio-tech (Shanghai, China). The analysis of pathway enrichment was compared to KEGG database.
Results
DZNep inhibits cell proliferation differentially with the EZH2-specific inhibitor GSK343 in HepG2 cells
DZNep is a carbocyclic analog of adenosine and was initially synthesized as an inhibitor of SAHH, 12 while recently it was reported to selectively induce apoptosis by preferentially targeting EZH2, the catalytic subunit of Polycomb-repressive complex 2 (PRC2), and the HMT responsible for H3K27me3. 13 Given DZNep involves in the inhibition of EZH2 through an indirectly manner (Figure 1(a)), we presume that it may have a more direct or acute biological effect on tumor cells than just reactivating tumor suppressor genes silenced by EZH2. Therefore, we first compared the pharmaceutical effects on HepG2 cells between DZNep and GSK343, a specific inhibitor directly targeting EZH2. 14 The results revealed that the two drugs affected the cell growth in a differential manner (Figure 1(b)).
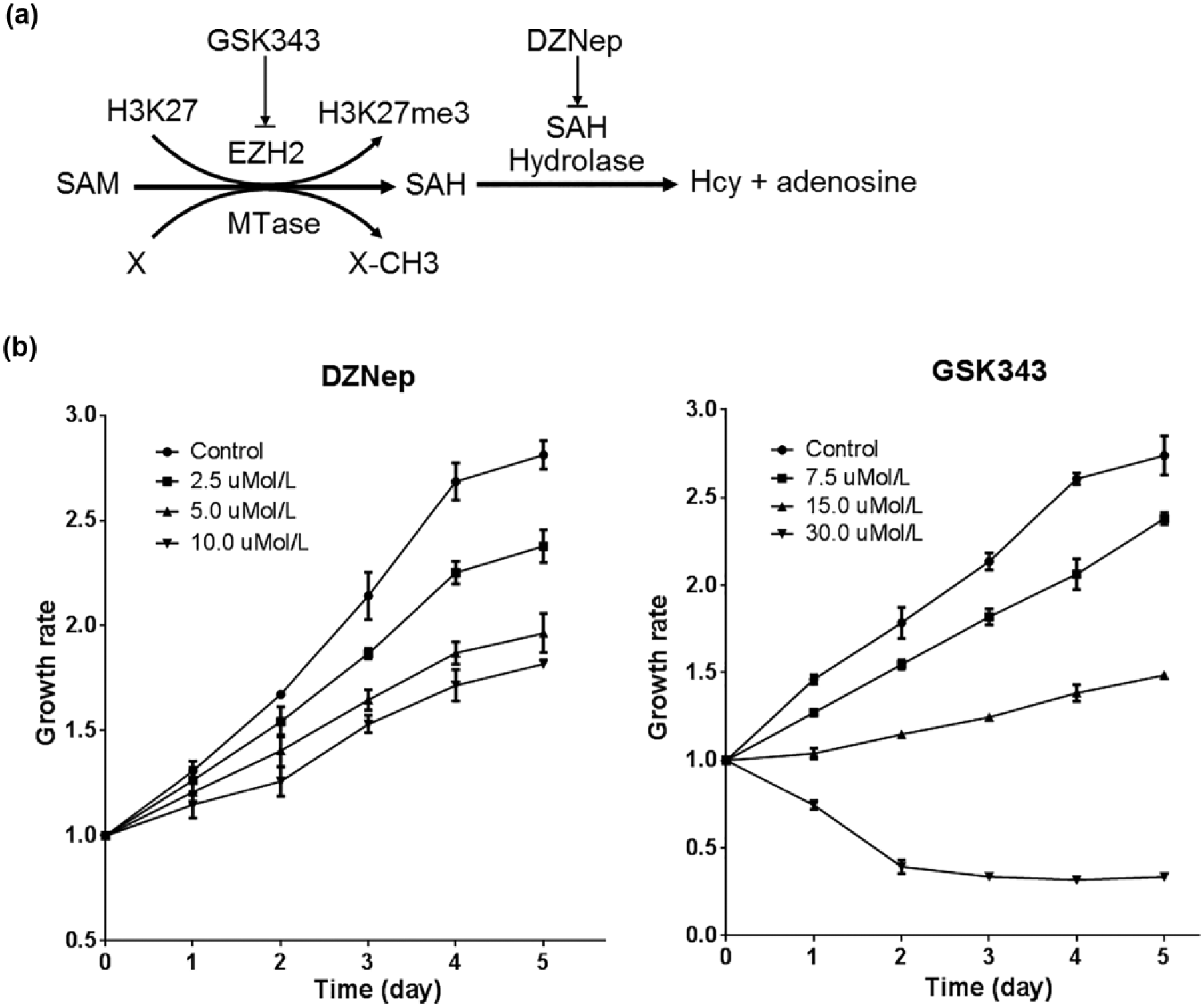
Differential inhibition to cell proliferation by DZNep and GSK343 in HepG2 cells. (a) Schematic of pharmacological targets for DZNep and GSK343. DZNep causes SAH accumulation by inhibiting SAH hydrolase; the elevated SAH potentially inhibits EZH2 and other methyl-transfer reactions (X to X-CH3). (b) Proliferation curves of DZNep and GSK343-treated cells.
First, DZNep inhibited cell growth in a mild way not directly proportional to its dose. DZNep at the concentration of 2.5 µmol/L suppressed the cell growth with an approximate 60% inhibition rate, but such quadrupled concentration (10.0 µmol/L) did not inhibit the growth further. In contrast, GSK343 inhibited cell growth in a dramatic manner. Second, different from GSK343’s obvious cell death effects, DZNep inhibited cell proliferation prominently via the induction of growth arrest. In particular, no more remarkable detached dead cells could be observed after refreshing culture medium after 3 days in DZNep-treated cells. Finally, to confirm that the intracellular SAH was accumulated during DZNep treatment, we qualitatively detected SAM and SAH in HepG2 cells by high-performance liquid chromatography (HPLC). The results revealed that SAH accumulated in DZNep-treated cells while it is removed in GSK343-treated ones (Supporting Information Figure S1).
DZNep causes morphological changes and senescence-like phenomena
In addition to the proliferation inhibition, morphological change was another DZNep’s biological effect on HepG2 cells. Since day 6, DZNep-treated cells still kept viable but began to gradually display enlarged, flattened morphology and increased granularity in cytoplasm. Such morphological alterations appeared extremely prominent on day 10. These senescence-like phenomena were confirmed by staining of senescence-associated β-galactosidase (SA-β-gal), a reliable biomarker for cellular senescence in vitro (Figure 2(a)). To examine whether such abnormal morphological changes were irreversible, after 10 days’ exposure to DZNep, the cells were passaged and cultured for another 7 days with or without the drug. It was demonstrated that withdrawal of DZNep could not improve the cell status with regard to cell morphology (Figure 2(b)). These observations suggested that DZNep-induced proliferation inhibition was not simply a result of transient growth arrest but was caused by permanent senescence.

Senescence-like characteristics of HepG2 cells upon 5.0 µmol/L DZNep. (a) On day 3, the cells appeared normal in morphology; on day 6, the cells displayed senescence-like morphologies and positive SA-β-gal staining in green (observed at magnification of 400×). (b) After 10-day DZNep treatment, withdrawal of drug could not increase cell density (100×) or reverse the senescence-like morphologies (200×).
DZNep induces prolonged G2/M arrest and enhances expression of cyclin-dependent kinase inhibitors p16 and p21
To investigate the mechanism underlining DZNep-induced senescence-like phenomena, the cell cycle was analyzed by flow cytometry. DZNep retained the cell cycle at G2/M phase on day 6 (Figure 3(a)). Since enhanced G2/M cell cycle checkpoint suggests a DDR triggered by genotoxicity, the expression of three checkpoint inhibitors (p16, p21, and p27) of DDR was further determined. As expected, the expression of p16 and p21 was significantly increased on the third day after exposure to the drug (Figure 3(b) and (c)). Therefore, the prolonged DNA damage checkpoints indicated that DZNep might induce a persistent or even permanent cell arrest through an enhanced DDR pathway.
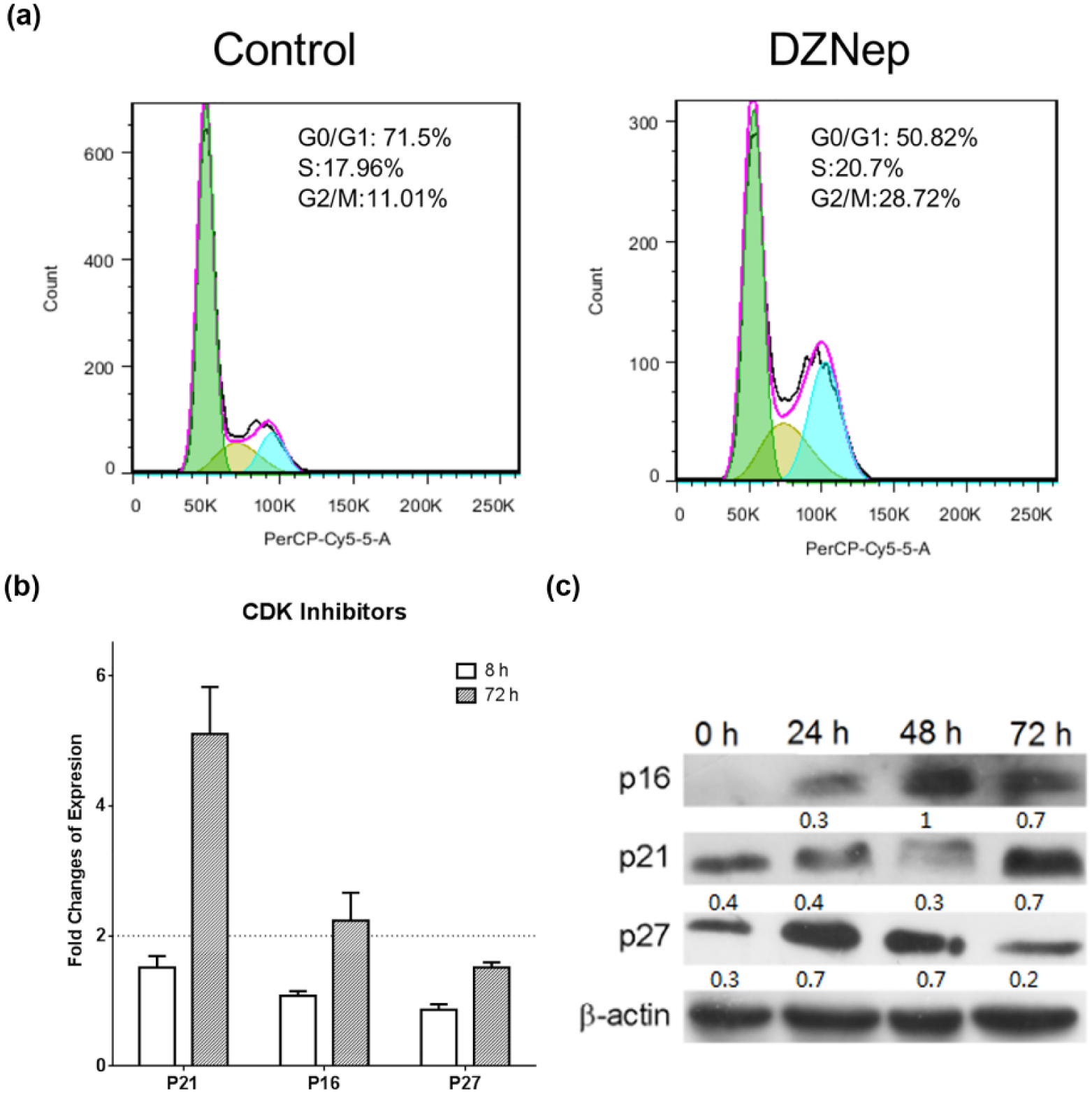
(a) Prolonged cycle arrest induced by 5.0 µmol/L DZNep. DZNep retained the cell cycle at G2/M phase on day 6. (b) mRNA expression of checkpoint inhibitors (p16, p21) upon DZNep treatment for 8 and 72 h. (c) Protein expression of checkpoint inhibitors (p16, p21) increased upon DZNep treatment on day 3. Quantification of the protein blot was performed by Image-Pro Plus (v6.0).
DZNep causes p53 stabilization but in a mild manner
Tumor suppressor p53 plays a pivotal role in sensing and responding to cellular stresses. 15 To verify that DZNep-induced senescence-like phenomena were mediated by p53 as a part of its response to genotoxic stress, the expression of p53 was examined. Western blot showed that the protein level was elevated on the third day upon DZNep treatment (Figure 4(a)).
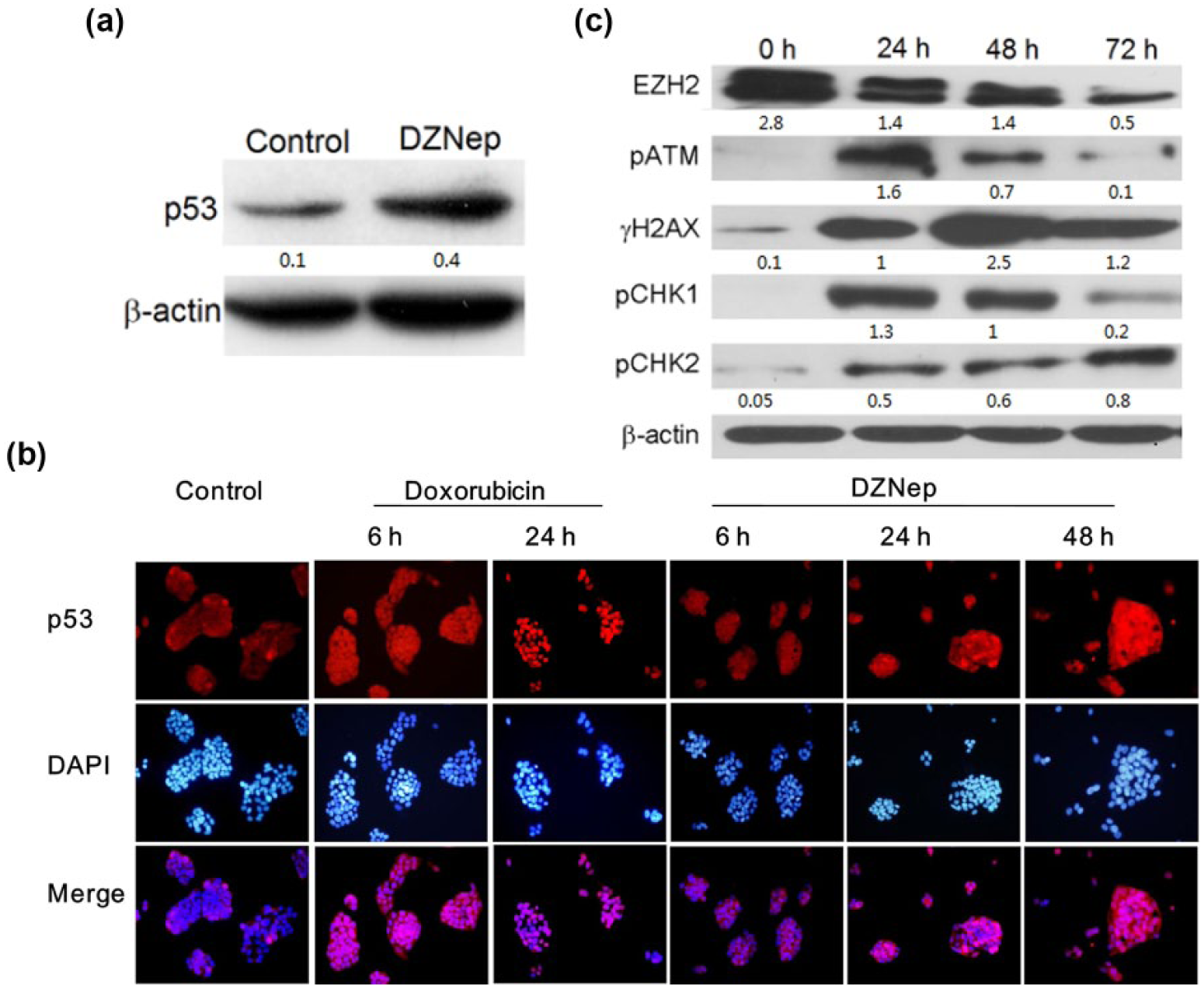
(a) p53 activation and DDR caused by 5.0 µmol/L DZNep. Expression of p53 protein elevated after 3-day treatment. (b) Doxorubicin strongly induced p53 stabilization and nuclearization after 6 h, while DZNep causes p53 accumulation gradually until 72 h. (c) Signaling proteins of DNA damage response were activated in the phosphorylation forms on the first day upon DZNep treatment, meanwhile the level of EZH2 decreased gradually along with drug treatment.
Under the stressed conditions, the activation of p53 needs to displace from the negative regulators (MDM2/MDM4) and subsequently import into nucleus. 16 To further confirm that DZNep-induced senescence was p53-dependent, we examined p53 translocation from cytoplasm to nuclear. Immunofluorescence analysis showed that nuclear p53 accumulation upon DZNep treatment progressively elevated to a moderate level till 72 h. In comparison, doxorubicin, a potent drug to generate double-strand breaks (DSBs) on DNA, strongly induced p53 stabilization and nuclear accumulation in 24 h (Figure 4(b)). Therefore, DZNep promoted the p53 stabilization in a mild and chronic manner.
DZNep triggers DDR as an early event in its pharmaceutical effects
To further confirm that DZNep resulted in senescence through DDR pathway, a panel of upstream signaling in DDR was analyzed by western blot (Figure 4(c)). Of the DDR signaling cascade, the protein kinase ATM is an apical activator and it transduces the alarm of DNA DSBs to downstream effectors. Autophosphorylation at Ser1981 is a hallmark of ATM activation. Notably, DZNep strongly activated the ATM in 24 h and then began to attenuate gradually. This indicated that ATM-mediated DDR was an early event in DZNep-induced senescence.
The activated ATM phosphorylates the histone variant H2AX on Ser139 (γH2AX), which is required for amplifying the DDR signaling and acts as a sensitive marker for DSBs. Upon ATM activation, γH2AX was detected at 24 h after DZNep treatment, and notably it continued to augment till 72 h. Similar to the above observation that p53 gradually stabilized during 72 h, this result implied a chronic but persistent DDR upon DZNep treatment.
Checkpoint kinases (CHK1 and CHK2) are downstream mediators of DDR signaling. CHK2 is mainly phosphorylated by ATM and can activate or stabilize several key effectors including p53. In agreement with the previous results for p53 and γH2AX, the level of pCHK2 also increased in a mild manner. Conversely, the level of pCHK1 dramatically increased in 24 h and attenuated thereafter. This hinted that the pathways of ATM-CHK2 and ATR-CHK1 may play different roles in the process of DDR.
Finally, in support of the notion that EZH2 is degraded upon DZNep in a proteasome-dependent way,17,18 our results also confirmed that DZNep effectively reduced EZH2 protein in 72 h. The depletion of this core component of PRC2 implied the disruption of repressive marker H3K27me3 on chromatin.
DZNep triggers senescence-associated inflammatory cytokine or chemokine secretion
Although transient activation of p53 in G2/M phase is sufficient to induce senescence together with p21 induction, 19 we wonder why the DDR-dependent cell fate in our experiments was preferably subject to senescence. To seek those factors specifically contributing to cellular senescence at the downstream of p53, we turned to the senescence-associated secretory phenotype (SASP) possibly elicited by DZNep. SASP includes multiple secreted inflammatory cytokines that reinforce growth arrest of senescent cells through their ability to boost the DDR.
We analyzed mRNA expression in a panel of inflammatory signals (IL1A, IL6, IL8, CXCR2, IGFBP7) frequently upregulated in senescence cells, together with two downstream effectors (PUMA and BIM) of p53-mediated apoptosis pathway. Remarkably, inflammatory factors CXCR2, IGFBP7, and IL8 were dramatically increased after 72 h upon exposure to DZNep. There was no significant upregulation for BIM and even supersession for PUMA (Figure 5(a)). These results implied that DZNep induced cellular senescence via DDR reinforced by SASP.

Genes related to SASP, histone, and DNA repair involving in DZNep-triggered DDR. DZNep did not affect mRNA level for all inflammatory signaling factors after 8-h treatment, but triggered a dramatic increase for CXCR2 (24.8 ± 2.4-fold, n = 3), IGFBP7 (16.0 ± 0.9-fold, n = 3), and IL8 (4.0 ± 0.5-fold, n = 3) after 72 h. The mRNA level of apoptotic effectors PUMA and BIM did not increase. No expression of IL6 was detected in either control or drug-treated cells. (a) The dash line denotes twofolds of expression increase. (b) The number of genes affected more than twofolds in expression upon DZNep and GSK343 treatment compared with the control. (c) The analysis of pathway enrichment revealed 10 mostly affected pathways by DZNep treatment. (d) RT-PCR validation of microarray data by screening a group of selected genes potentially predicting drug response to DZNep. (e) Schematic for the role of DZNep in disrupting the epigenome status of established heterochromatin and thereby enhancing the DDR to endogenous genotoxicity or DNA-damaging agents.
Genes involved in histone variants and DNA repair upregulate upon DZNep treatment
To further investigate the genes involved in DDR upon drug treatment, HepG2 cells were treated with DZNep and GSK343 for 72 h, and the whole-genome expression profiles were analyzed by microarray. First, among the genes with over two fold changes in expression, there was only a small part (14.3% for up- and 17.3% for downregulation) simultaneously occurring in both DZNep and GSK343 groups (Figure 5(b)). This was consistent with their different pharmacologic effects on HepG2 cells.
Second, enrichment analysis revealed 10 most related pathways upregulated or activated in DZNep-treated cells (Supporting Information Table S1). Impressively, the first two actually contained one same group of histone H2A, H2B, and histone H3 genes, despite the pathways being annotated as “alcoholism” and “systemic lupus erythematosus,” respectively (Figure 5(c)). These results strongly implied a nucleosome remodeling in response to the perturbation of chromatin due to DZNep treatment. In addition, in support of the previous result that DZNep-triggered senescence was an outcome of ATM–CHK2–p53 axis activation, one group of p53-associated genes were identified as well in the pathway enrichment analysis.
Finally, to validate the microarray results and screen the candidate biomarkers for DZNep response, 15 upregulated and 2 downregulated genes in microarray analysis were further determined by RT-PCR. The results confirmed the reliability of microarray data, and more importantly, at least three known genes (ALDH3A, 20 TRIM29, 21 and TP53I3 22 ) closely associated with DNA repair were identified to upregulate significantly in DZNep-treated cells. In radio- or chemotherapy, the activation of DNA repair pathway may predicate a poor prognosis due to attenuated DDR signaling. This can also partly explain why DZNep triggered senescence but not apoptosis. Therefore, targeting these genes involved in DNA repair pathways before the onset of senescence may represent an intervention to enhance the therapeutic effects of DZNep or other genotoxic anticancer agents.
Discussion
In human cancers, the levels of heterochromatin markers are higher than in normal tissues. In particular, SAHF are preferentially formed following oncogene activation and represent a barrier to DDR signaling.7,8 Pharmacological perturbation of heterochromatin in oncogene-expressing cells increases DDR signaling and improves therapeutic efficacy. 7 Given the close link between the metabolic cycles of methionine and HMTs, 23 in this study we explored the pharmacologic inhibitor of SAHH as a feasible intervention to perturb the chromatin status by modulating the balance of SAM and SAH metabolism, and demonstrated DZNep-elicited cellular senescence through a sensitized DDR pathway mediated by ATM in cancer cells. In brief, DZNep disturbs the established epigenome and enhances the sensitivity of hepatoma cells to endogenous genotoxic stress.
There are at least two means whereby DZNep affects the chromatin status of cancer cells. First, DZNep causes SAH accumulation and decreases the ratio of SAM/SAH. 24 Now increasing evidences have shown that such perturbation in methionine metabolism is sufficient to alert the epigenetic status of chromatin.10,25 Histone marks of H3K4me3, H3K9me3, and H3K27me3 are each known to have substantial roles in defining chromatin states. It has been demonstrated that all these markers can be globally decreased by methionine restriction or DZNep treatment. 10 Second, DZNep evokes recent focus for its reported specific disruption of EZH2-mediated H3K27Me3. 13 Such preference was attributed to the proteasome-dependent degradation of EZH2.17,18 EZH2 plays an essential role in the maintenance of both the proliferative and self-renewal capacity of hepatic stem/progenitor cells and the full execution of their differentiation. 26 In our result, the gradual decrease in EZH2 upon DZNep treatment was also observed in HepG2 cells. Together with other clue, the depletion of PRC2 by DZNep may have earlier and more prominent effects on the chromatin structure than the transcriptional activation of specific tumor suppressor genes. 27 The sensitivity and specificity of EZH2 for hepatocellular carcinoma were reported to be 95.8% and 97.8%, respectively. 28 It is worthy to test in more cell lines and tumor model to evaluate whether EZH2 could be a promising target by DZNep.
Chromatin status affects initiation and amplification of DDR signaling as well as accompanied DNA repair. First, as an early event in cellular responses to genotoxic stress, ATM activation can result from the changes in chromatin structure without DNA strand breaks. 29 Second, the spread of γH2AX facilitates the magnification of DDR signaling to stress, while it can be restricted from heterochromatin. 30 In addition, an increasing list of histone methylations (e.g. H3K20me1/2/3, 31 H3K36me2/3,32,33 and H3K79me2 34 ) is found to involve in DNA repair by directing DDR mediator to recruit at the sites of DSBs.
In summary, the close link between methionine metabolism, epigenetic landscape, and DDR provides the rationale for pharmacologically perturbing the epigenome status of established heterochromatin to enhance the sensitivity of cancer cells to DDR signaling (Figure 5(e)). In this study, therapy-induced senescence by DZNep exemplifies a mild cellular response to endogenous genotoxic stress (e.g. activated oncogenes, replicative stress, or increased reactive oxygen species (ROS)). In combination with DNA-damaging agents or inhibitors to DNA repair, such chromatin-modifying drugs would further improve cancer treatment outcomes.
Footnotes
Acknowledgements
G.W. and N.W. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Municipal Key Project of Tianjin Health Bureau (grant nos 12KG107 and 12KG108).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.