
Abstract
Pancreatic cancer has one of the lowest survival rates of all cancers. The mechanism underlying chemo-resistance of pancreatic cancer is not well understood. Our previous article reported that small molecule YM155 induced apoptosis in pancreatic cancer cells via activation of death receptor 5. In this study, we aim to continuously address death receptor 5–mediated apoptosis in chemo-resistant pancreatic carcinoma. We found that in comparison to paired pancreatic cancer tissues and adjacent normal tissues, five of the six cancer tissues had downregulated death receptor 5 and upregulated Bcl-xL. Mono treatment with lexatumumab was not sufficient to induce apoptosis in pancreatic cancer cells, whereas focal adhesion kinase inhibitor PF573228 significantly sensitized lexatumumab-induced apoptosis. Western blotting analysis revealed that lexatumumab and PF573228 combination treatment increased death receptor 5 but decreased Bcl-xL expression. Interestingly, pre-treatment with Bcl-xL inhibitor ABT263 reversed the insensitivity of panc-1 cells to lexatumumab or PF573228-induced apoptosis. Specific small interfering RNA-mediated gene silencing of Bcl-xL effectively sensitized pancreatic cancer cells to lexatumumab or PF573228-induced apoptosis. Furthermore, lexatumumab and PF573228 combination was shown to exhibit significant xenograft pancreatic tumor growth inhibition in SCID mice. Our data provide fundamental evidence to support the notion that lexatumumab and PF573228 co-treatment could be a potentially effective regime for patients with pancreatic cancer.
Introduction
Pancreatic cancer is terminal in most cases with less than 5% survival rate.1,2 To date, surgical resection is the most effective treatment option to cure this malignant tumor. Most cases are detected at a late stage and are unresectable due to metastasis. The majority of pancreatic cancer patients do not survive more than 1 year post diagnosis.3,4 Unfortunately, pancreatic cancer demonstrates a broad resistance to current conventional chemotherapeutic agents, which are administered with the aim of inducing tumor cell apoptosis.5,6 Therefore, new chemotherapeutic strategies that sensitize pancreatic cancer cells to secondary drug treatment are required for success.
Focal adhesion kinase (FAK), a protein tyrosine kinase, is able to mediate signal transduction from extracellular matrix (ECM) to cells. It has been reported that FAK is involved in cell proliferation, survival, motility, and invasion via interaction with integrin and growth factor receptors.7,8 Overwhelming evidence has been compiled that identifies FAK as a key molecule conferring chemotherapeutic resistance to pancreatic cancers, and resistance is conferred by enhancing anti-apoptotic properties in tumor cells.9–11 A number of specific inhibitors of FAK have been developed. 12 Of all, PF573228 (PF) is well documented and characterized. 13 PF is able to inhibit FAK activity by blocking the phosphorylation of FAKTyr397 and has been shown to induce apoptosis or growth inhibition in prostate metastatic (PC3) cells, human ovarian adenocarcinoma (SKOV-3) cells, gemcitabine-resistant pancreatic cancer (L3.6p1) cells, pancreatic cancer (F-G) cells, and canine kidney epithelial (MDCK) cells.14,15 However, the anti-tumor effects of PF on chemo-resistant pancreatic cancer cells are poorly known.
Lexatumumab (Lexa/HGS-ETR2) is a fully humanized agonistic monoclonal antibody (mAb) to the tumor necrosis factor–related apoptosis-inducing ligand receptor 2 (TRAIL-R2/death receptor 5 (DR5)).16,17 It has been shown that Lexa specifically binds to DR5 and induces apoptosis in a number of tumor cell lines, including renal cell carcinoma (RCC), human myeloma cell lines (HMCLs), and malignant pleural mesothelioma (MPM). 18 Our previous data have shown that Lexa and cycloheximide (CHX) in dual-combination treatment can induce apoptosis in hepatocellular carcinoma (HCC) cells via the upregulation reactive oxygen species (ROS) levels, without generating apoptotic effects in normal liver cells. 19 The beneficial synergy between Lexa and PF chemotherapy in pancreatic cancer has not been determined to date.
The purpose of this study is to investigate how to use PF or Lexa to induce apoptosis in chemo-resistant pancreatic cancer cell lines. We found deregulated expression of DR5 and Bcl-xL in a majority of pancreatic cancer tissues when compared with adjacent normal pancreatic tissues. Our results show that the combination of Lexa and PF induces robust apoptosis in chemo-resistant pancreatic cancer cells, whereas monotreatment with either Lexa or PF has no apoptotic toxicity. We show that inhibition of the anti-apoptotic Bcl-2 family protein Bcl-xL can sensitize either Lexa or PF to induce apoptosis in pancreatic cancer cells.
Materials and methods
Materials
ABT263 and PF were purchased from Selleck Chemical (Houston, TX); anti-cleaved Caspase 8, anti-cleaved Caspase 3, anti-Bcl-xL, anti-Bcl-2, anti-Bax, anti-Bad, anti-FAK, anti-Mcl-1, and anti-DR5 primary antibodies were obtained from Cell Signaling Technology (Beverly, MA); the Annexin-V apoptosis detection kit was obtained from BD Bioscience (San Diego, CA). Hoechst 33258 and anti-β-actin were obtained from Sigma (St Louis, MO); goat anti-rabbit horseradish peroxidase (HRP)–conjugated secondary antibody and goat anti-mouse secondary antibody conjugated with HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Lexatumumab was kindly provided by Human Genome Science Inc. (Rockville, MD).
Cell culture and reagents
Human pancreatic cancer cell lines, Panc-1 and AsPc-1, were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (Sigma) and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin) at 37°C in 5% CO2.
Hoechst staining assay
Apoptosis was determined through nuclear morphology change. After treatment with different stimuli, cells were stained with Hoechst 33258 at 37°C for 10 min. Cell nuclear condensation was detected using an Olympus fluorescent microscope (Tokyo, Japan). Apoptotic cell death ratio was assessed by the counting of apoptotic cells with condensed nuclei in six to eight randomly selected areas.
Fluorescence-activated cell sorting assay
Apoptosis was measured using the Annexin-V detection kit according to the manufacturer’s instructions. Flow cytometric analysis was performed to monitor the green fluorescence of the fluorescein isothiocyanate (FITC)–conjugated Annexin-V and the red fluorescence of DNA-bound propidium iodide (PI). All data were analyzed using Cell Quest software (BD).
Western blotting assays
Cells were harvested and washed twice with sterile cold phosphate-buffered saline (PBS). The cell pellets were resuspended in lysis buffer containing Nonidet P-40 (Cell Signaling Technology) and incubated on ice for 30 min. After centrifugation at 12,000g at 4°C for 15 min, the supernatant was transferred to a new tube and the protein concentration was determined. Equivalent samples (20 µg of protein) were subjected to sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) on 12% gels. The proteins were transferred to nitrocellulose membranes and probed with the indicated primary antibodies, followed by the appropriate secondary antibodies conjugated with HRP. Immunoreactive bands were detected using enhanced chemiluminescence (ECL; Pierce, Rockford, IL). The molecular sizes of the proteins detected were determined by comparison with prestained protein markers (Bio-Rad, Hercules, CA).
Small interfering RNA knockdown
Small interfering RNA (siRNA) knockdown was performed as previously described. 20 Smart-pool pre-designed siRNA duplexes targeted against human Bcl-xL messenger RNA (mRNA) were from Cell Signaling Technology. Cells were plated at a density of 1 × 105 cells/well in six-well plates (BD Bioscience). Next day, cells were transfected with 100 nM siRNA duplex mixtures for 24 h in the presence of lipo-fectamine RNAiMax (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. A non-specific random siRNA (Cell Signaling Technology) was also transfected at the same concentration as control.
Tumor growth inhibition test
Mice were managed humanely in line with the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” (NIH publication 86-23, revised 1985). Eight-week-old male SCID mice were divided into two groups. Each mouse (five per group) was inoculated subcutaneously at a dorsal flank site with 1 × 107 cells in PBS. Tumor volume was measured with calipers once every 3 days. When tumor volume reached 100 mm 3 , mice were administrated with PBS or Lexa + PF via tail vein injection every other day for a total of 15 days. The effect of drug treatment on tumor growth was analyzed statistically with individual group comparison and evaluated by Student’s paired t-test by SPSS 15.0 (Chicago, IL).
Statistical analysis
The experiments were repeated in triplicate with consistent results, and representative findings are shown. Statistical analysis was performed using Student’s t-test analysis, with p values <0.05 considered significant.
Results
Dysregulation protein expression in pancreatic cancer tissues
To verify the mechanism underlying the resistance of pancreatic cancer cells to chemotherapy, paired malignant pancreatic tissues and adjacent relative normal pancreatic tissues were collected. We compared protein expressions between paired pancreatic cancer and adjacent normal pancreatic tissues. Paired tissues were processed for western blotting to analyze protein levels of FAK, DR5, Bcl-xL, Bcl-2, Mcl-1, and Bax. As shown in Figure 1, our data revealed that 83% (five of six) of malignant tissues had lower expression of DR5 when compared with adjacent normal tissues. Malignant tissues had higher expression of Bcl-xL and Bax (83%, five of six) than adjacent normal tissues, while the other two proteins Bcl-2 and Mcl-1 did not show apparent difference between normal and cancer tissues. We observed that FAK was elevated in 50% (3three of six) cancer tissues. These data suggest that DR5 and Bcl-xL were dysregulated and might be crucial in malignant pancreas tissues.
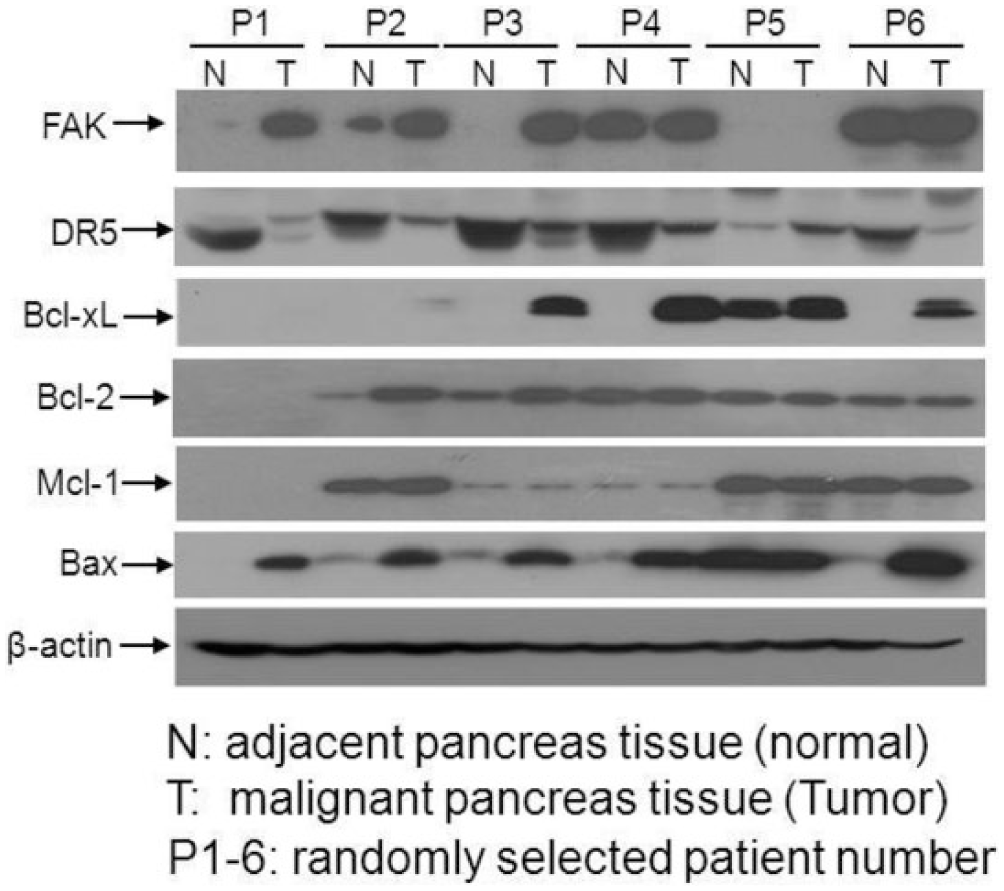
Dysregulated protein expression in malignant pancreas tissues. Paired malignant tissues and normal adjacent normal tissues were collected from patients with pancreatic cancer. Experiments were performed to assess the protein levels of FAK, DR5, Bcl-xL, Bcl-2, Mcl-1, and Bax. Tissues were homogenized in lysis buffer, and lysates were prepared for western blotting analysis. FAK, DR5, Bcl-xL, Bcl-2, Mcl-1, and Bax were examined with specific antibodies. β-Actin protein levels were assessed as loading controls for equal total protein amounts. Representative bands are shown, and every experiment was repeated three times.
PF sensitizes Lexa to induce apoptosis in pancreatic cancer cells
It has been reported that both Panc-1 and AsPc-1 cells are resistant to TRAIL-induced apoptosis.21–23 To determine whether the chemo-resistance of Panc-1 cells and AsPc-1 cells is associated with DR5 dysregulated lower levels, cell lines were treated with dimethyl sulfoxide (DMSO) (Control), Lexa (1 µg/mL), PF (10 µM), or both Lexa and PF for 24 h (Panc-1 cells) or 48 h (AsPC-1 cells). Nuclear condensation or DNA fragmentation assays were performed to measure apoptosis. Our data indicated that single treatment with either Lexa or PF did not induce apoptosis in Panc-1 cells or AsPc-1 cells. Interestingly, combination treatment triggered a robust apoptotic response in both cell lines as determined by nuclear condensation detected in a majority of cells (Figure 2(a)). To further confirm the apoptotic effects of Lexa and PF combination treatment, Panc-1 cells were treated with Lexa, PF, Lexa and PF or not and fluorescence-activated cell sorting (FACS) based on Annexin V-PI double staining was performed to assess apoptosis ratio. Consistent with the nuclear condensation results, the combination of Lexa and PF induced cell death in >70% of pancreatic cancer cells (Figure 2(b)).To assess the intracellular apoptotic events induced by combined treatment, Caspase 8 and Caspase 3 activation was examined by western blotting analysis to show cleaved bands; our data indicated that only combination treatment triggered Caspase 8 and Caspase 3 activation as determined by detection of Caspase cleavage products (Figure 2(c)). These data suggest that Lexa-induced apoptosis can be activated in the presence of FAK inhibition by PF in resistant pancreatic cancer cells.

PF and Lexa synergistically induce apoptosis in pancreatic cancer cells. (a) Panc-1 and AsPc-1 cells were treated with DMSO (Control), Lexa (1 µg/mL), PF (10 µM), and Lexa (1 µg/mL) + PF (10 µM) for 24 h (Panc-1 cells) or 48 h (AsPc-1 cells). Nuclear condensation was assessed by Hoechst 33285 staining to show apoptotic nuclei as described in section “Materials and methods” (representative apoptotic cells are marked by white arrows). (b) Panc-1 cells were treated with DMSO (Control), Lexa (1 µg/mL), PF (10 µM), or Lexa (1 µg/mL) + PF (10 µM) for 24 h. Apoptosis was evaluated through FACS analysis based on Annexin-V and PI double staining using the Annexin-V assay kit. The percentage of apoptotic cells was characterized as those that stained with Annexin-V. The data represented show the mean values of three independent experiments (*p < 0.05). (c) Panc-1 cells or AsPc-1 treated with indicated conditions for 24 or 48 h were harvested, and cell lysates were prepared and subjected to western blotting analysis. Caspase 8 and Caspase 3 activation was evaluated through detection of cleaved bands with specific antibodies. β-Actin protein levels were assessed as loading controls for equal total protein amounts. Representative bands are shown, and every experiment was repeated three times.
Protein expression in PF and Lexa-treated pancreatic cancer cells
To determine the molecular pathways influencing the sensitivity of pancreatic cancer cells to the combination treatment, we examined protein expression of key components of the intrinsic and extrinsic apoptosis pathways. Western blotting analysis demonstrated that the addition of PF increased DR5 expression levels by up to 10-fold (Figure 3). We observed that the combination treatment (PF 10 µM and Lexa 1 µg/mL) decreased the expression of Bcl-xL protein by up to 7-fold (Figure 3). The anti-apoptotic Bcl-2 and pro-apoptotic molecules Bax and Bad did not show any significant difference (Figure 3). These data suggest that the activity of PF and Lexa may be mediated by activating DR5 and decreasing Bcl-xL.

Protein expression in Lexa, PF, or Lexa and PF-treated pancreatic cancer cells. Panc-1 cells were treated with or without Lexa (1 µg/mL), PF (10 µM), or pre-treated with PF (10 µM) followed by Lexa (1 µg/mL) for up to 24 h. Cells were harvested and cell lysates were prepared for western blotting. DR5, Bcl-xL, Bcl-2, Bax, and Bad expressions were detected with specific antibodies. β-Actin protein levels were assessed as loading controls for equal total protein amounts. Representative bands are shown, and every experiment was repeated three times. The relative intensity of immunoblot bands was analyzed by ImageJ software.
Bcl-2 inhibitor ABT263 sensitizes Lexa- and PF-induced apoptosis
Bcl-xL inhibition could sensitize Survivin inhibitor YM155 to induce apoptosis in resistant HCC cells. 24 In this study, we found that Bcl-xL was elevated in malignant tissues and downregulated by PF and Lexa co-treatment-induced apoptosis. Therefore, to verify the roles of Bcl-xL in chemotherapy, we first used ABT263 to inhibit Bcl-xL activity. Cells were pre-treated with sub-lethal ABT263 followed by Lexa or PF. As shown in Figure 4(a) and (b), Lexa or PF induced significant apoptosis in the presence of ABT263. These data suggest that Bcl-xL inhibition is very crucial to cause sensitivity converse to treatment with either Lexa or PF.
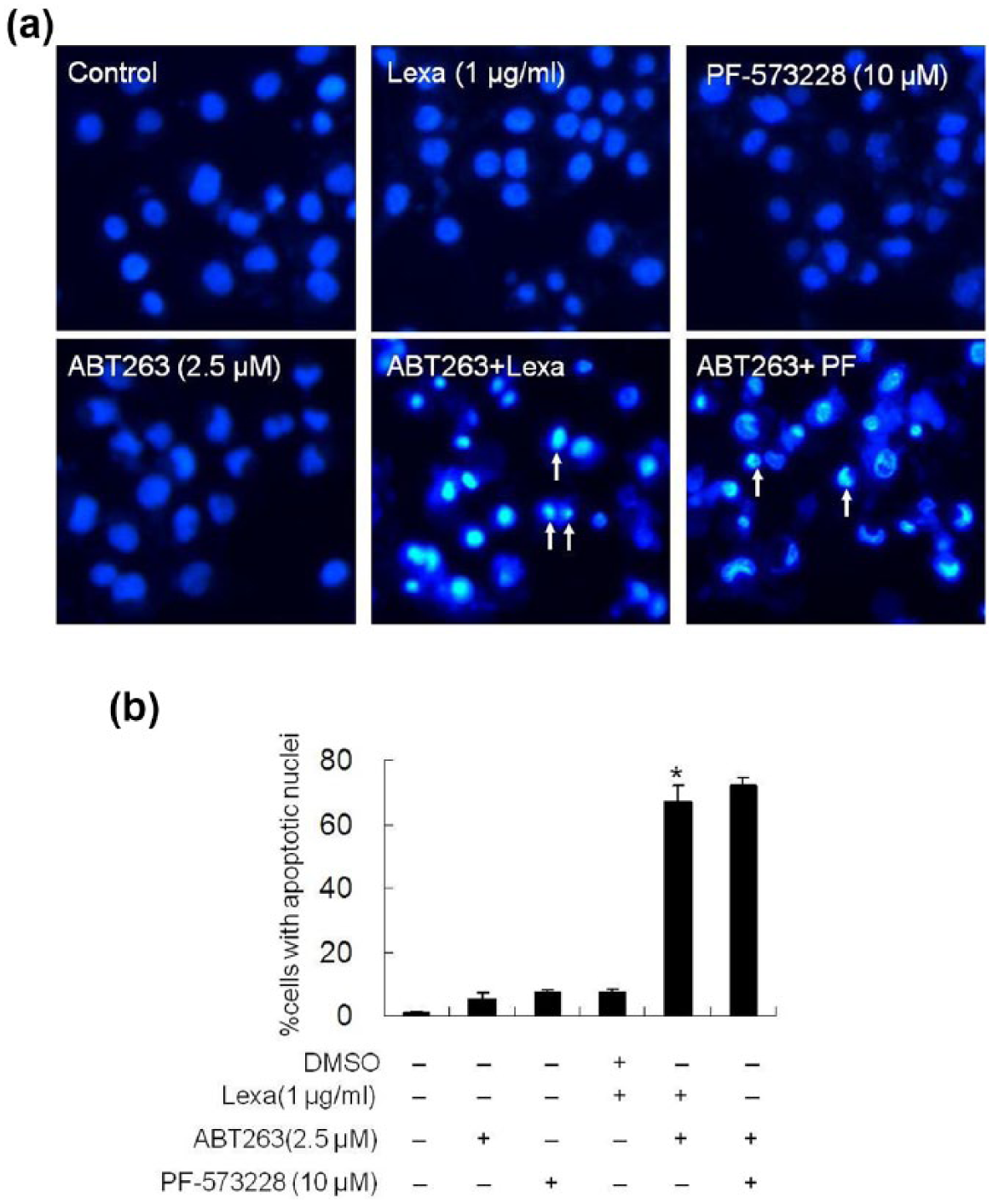
Bcl-xL inhibition by ABT263 enhances Lexa/PF-induced apoptosis. (a) Panc-1 cells were treated with DMSO (Control), Lexa (1 µg/mL), PF (10 µM), ABT263 (2.5 µM), or pre-treated with ABT263 (2.5 µM) followed by Lexa (1 µg/mL) or PF (10 µM) for 24 h. Cells were then stained with Hoechst 33258 to assay apoptotic cell death (representative apoptotic cells are marked with white arrows). (b) Apoptotic ratio in Figure 4(a) was determined by counting cells with apoptotic nuclei as described in section “Materials and methods.” Data represent the mean values of three independent experiments (*p < 0.05).
Bcl-xL knockdown sensitizes pancreatic cancer cells to Lexa/PF-induced apoptosis
To further ascertain the specific member, the Bcl-2 family regulating chemo-resistance, we used Bcl-xL-specific siRNA to knockdown its expression. Our results indicated that Bcl-xL siRNA-mediated gene silencing was highly efficient as compared to the control random siRNA (Figure 5(a)). To show that reduction in Bcl-xL expression is linked to chemo-resistance and sensitivity to apoptosis, Lexa or PF were used to treat cells harboring random siRNA or Bcl-xL-specific siRNA. Nuclear condensation assays demonstrated that Bcl-xL knockdown sensitized pancreatic cancer cells to both Lexa and PF to induce apoptosis (Figure 5(b) and (c)). To further verify that depletion of Bcl-xL increased Lexa- or PF-induced apoptosis, we assessed Caspase 8 and Caspase 3 cleavage activation. Western blotting analysis showed that upon Lexa or PF stimulation, significant cleaved Caspase 8 or Caspase 3 accumulated only in Bcl-xL gene silencing cells (Figure 5(d)). These data suggest that Bcl-xL activity inhibition is indeed essential for the loss of chemo-resistance in pancreatic cancer cells, enabling the induction of apoptosis by either Lexa or PF.
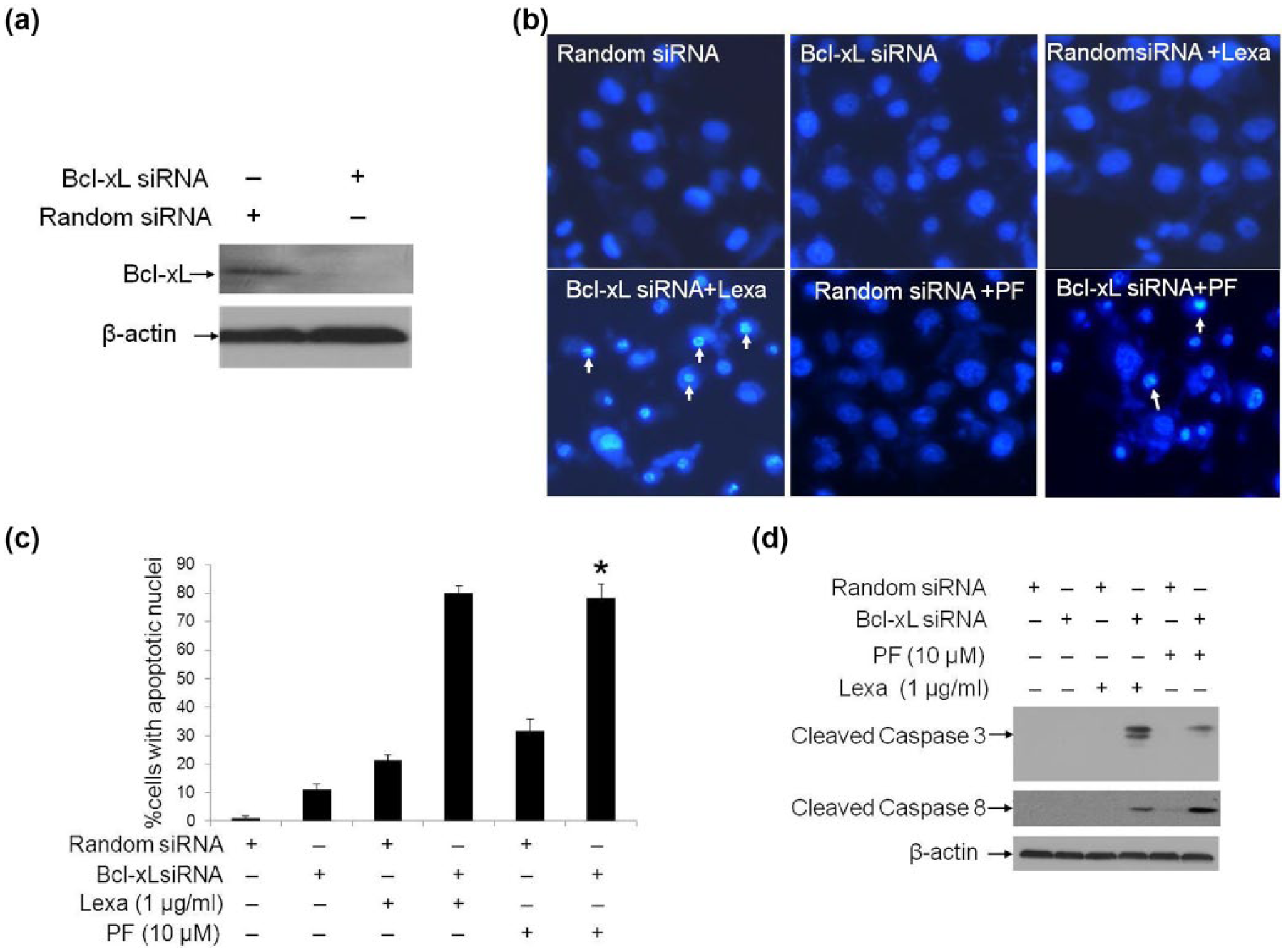
Bcl-xL knockdown sensitizes pancreatic cancer cells to Lexa/PF-induced apoptosis. (a) Panc-1 cells were transfected with synthesized random siRNA (as control) and Bcl-xL-specific siRNA, and after 48 h post-transfection cells were subjected to western blotting analysis with Bcl-xL specific polyclonal antibody. β-Actin protein levels were assessed as loading controls for equal total protein amounts. Representative bands are shown, and every experiment was repeated three times. (b) Panc-1 cells transfected with random siRNA or Bcl-xL siRNA were treated or not with Lexa (1 µg/mL) or PF (10 µM) for 24 h. Then cells were stained with Hoechst 33258 to test apoptotic cell death (representative apoptotic cells are marked with white arrows). (c) Apoptotic ratio in Figure 5(b) was determined by counting cells with apoptotic nuclei as described in section “Materials and methods.” Data represent the mean values of three independent experiments (*p < 0.05). (d) Panc-1 cells treated as in Figure 5(b) were harvested, and cell lysates were prepared and subjected to western blotting analysis. Caspase 8 and Caspase 3 activation was assessed by the detection of cleaved bands with specific antibodies recognizing cleaved Caspase 8 and Caspase 3. β-Actin protein levels were assessed as loading controls for equal total protein amounts. Representative bands are shown, and every experiment was repeated three times.
PF and Lexa combination inhibits tumor growth in vivo
To investigate the in vivo anticancer effects of PF and Lexa on pancreatic carcinoma, xenograft tumor derived from Panc-1 cells were established in SCID mice. Briefly, 10 mice were injected with Panc-1 cells to form xenograft tumors. One group (five mice) was treated with PBS buffer (control group) and the other group (five mice) was administrated with PF and Lexa together. The results showed that the control mice had developed much larger and more highly vascularized tumors than those mice treated with PF and Lexa (Figure 6(a)). There was a significant difference in tumor mass with at least a 3-fold decrease in PF and Lexa-treated mice when compared with untreated control mice (Figure 6(b)). To verify the involvement of DR5 upregulation and Bcl-xL downregulation in PF and Lexa combination-induced tumor growth inhibition, tumor tissues from control or treated group were processed for western blotting. Our data indicated that PF and Lexa co-treatment indeed was able to promote DR5 expression, while reduce Bcl-xL in five randomly paired tissues (Figure 6(c)). To verify Lexa and PF combination treatment induced apoptosis in xenograft tumor inhibition, apoptotic marker protein Caspase 3 cleavage activation was measured. In comparison to control group, we found that cleaved Caspase 3 was increased in five Lexa and PF co-treated tumor tissues, but not in control groups. These data suggest that the combination could effectively inhibit malignant pancreatic tumor growth in vivo by inducing apoptosis.
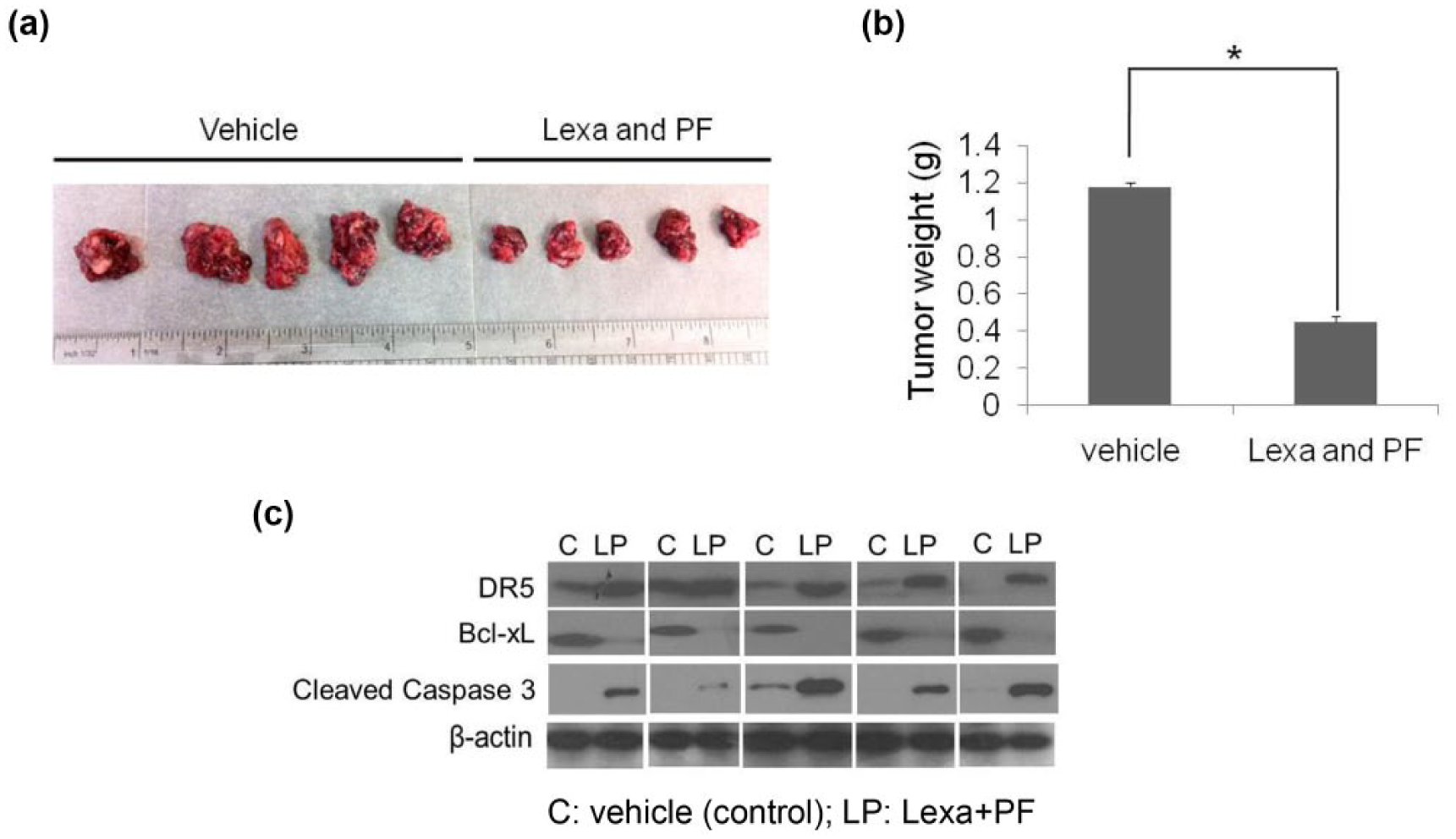
Lexa and PF induce tumor growth inhibition in vivo. (a) BALB/c SCID mice were inoculated subcutaneously with 1 × 107 Panc-1 cells in 100 µL PBS. The Panc-1 xenografts were treated with the combination of Lexa (10 mg/kg) and PF (100 mg/kg) every other day, i.v. injection until day 15. At the end point of the experiment, all the mice were sacrificed to take the tumors out. Pictures of excised tumors demonstrate larger tumors from untreated (control) mice and smaller tumors from Lexa and PF-co-treated mice. (b) Each tumor was weighed, and the mass of tumors from control and treated group was compared. Statistical analysis was performed to show inhibitory effects of Lexa and PF on the mass of implanted tumors (*p < 0.05). (c) Xenograft pancreatic tumors from untreated groups and Lexa and PF-treated groups were collected and lysates were prepared. Western blotting was performed to detect protein levels of DR5, Bcl-xL, and cleaved Caspase 3. β-Actin was set up as an equal protein loading control. The five pairs of tissues tested were randomly selected sample pairs, from untreated or Lexa and PF-treated tumor mice.
Discussion
In this study, we first provide evidence to show that DR5 activation induced by DR5 agonist Lexa could not promote apoptosis in pancreatic cancer cells. Significant apoptotic effects were observed when Lexa was used along with PF. Bcl-xL downregulation appears to be critical to sensitize PF and Lexa-induced apoptosis. This combination treatment of PF and Lexa exerts tumor growth inhibition in a xenograft pancreatic cancer mouse model.
It is well documented that pancreatic cancers are insensitive to current pancreatic cancer agents including gemcitabine, 25 TRAIL, 26 and a very promising FAK inhibitor PF.27–29 Reports from different research groups have argued that multidrug resistance–associated protein 2 (MDRP2), 30 IAP family proteins, 31 RAF/ERK, 32 Akt/mTOR, 33 heat-shock protein 27 (HSP27), 34 PKC, 35 Wnt/β-catenin, 36 and microRNAs 37 are involved in pancreatic cancer resistance. Therefore, it is important to define the intracellular molecular targets that may contribute to new therapies. In this study, we analyzed the proteins’ expression in paired pancreatic cancer tissue and adjacent normal pancreatic tissues. Interestingly, we found that in a small cohort of six cancer patients, five of the six pancreatic tumor tissues had a lower DR5 expression than the adjacent non-tumor pancreatic tissues, suggesting that DR5-mediated apoptosis pathway deregulation may be an important determinant in chemotherapy resistance of pancreatic cancer cells to TRAIL and contributes to the progression of metastasis. To rule out the possible involvement of DR5 in chemo-resistance of pancreatic cancer, we used Lexa, a mAb that specifically binds to DR5 receptor to activate apoptosis. Lexa is currently in phase I/II clinical trials in patients with various advanced malignancies. 38 Here, we found that Panc-1 and AsPc-1 cells are resistant to Lexa-induced apoptosis, suggesting that the DR5-mediated apoptosis signaling pathway does not function in these malignant cells. This is in accordance with previous reports that Panc-1 and AsPc-1 cells are not responsive to TRAIL-induced apoptosis and Panc-1 cells are resistant to Lexa.23,39 It has been reported that the majority of pancreatic cancers over-express FAK and FAK activation was able to abrogate TRAIL-induced apoptosis in a number of cancer types such as ovarian cancer 40 and colon cancer. 41 Consistently, our data also showed that 50% malignant tissues had elevated FAK (Figure 1). Therefore, to verify the involvement of FAK in the prevention of Lexa-induced apoptosis in pancreatic cancer cells, we performed experiments pre-treating pancreatic cancer cells with the FAK specific inhibitor PF; pre-treatment of cells was followed by treatment with Lexa. We found that sub-lethal doses of PF successfully reversed the insensitivity of Lexa, suggesting that FAK inhibition is indeed critical for DR5-mediated cell death in these pancreatic cancer cells. Interestingly, we also observed that treatment with PF increased the expression of DR5, making Lexa the optimal partner in this combination treatment, as Lexa induces apoptosis by binding to DR5 to initiate the intrinsic pathway. This is in accordance with our recent report that DR5 increase induced by YM155 is crucial for chemotherapy-induced apoptosis in Panc-1 cells. 42
In this study, we also noticed that five of the six malignant tissues exhibited much higher anti-apoptotic Bcl-xL expression, which may provide a possible explanation why these malignant cells are insensitive to chemotherapy such as Lexa or PF. We hypothesized that FAK inhibition may affect intracellular Bcl-2 family proteins and sensitize cancer cells to Lexa-induced apoptosis. As expected, we found distinct Bcl-xL protein decrease (7-fold) under the combination treatment (Lexa 1 µg/mL + PF 10 µM) suggesting Bcl-xL expression loss induces chemotherapeutic sensitivity in pancreatic cancer cells. To confirm the notion that Bcl-xL plays a key role in resistance of pancreatic cancer cells, a specific inhibitor ABT263 was used to block Bcl-2 family activity and then cells were treated with Lexa or PF. Surprisingly, we found that ABT263 effectively sensitized Panc-1 cells to undergo apoptosis upon treatment with either Lexa or PF. This is consistent with studies that ABT263 for the enhancement of TRAIL-mediated cell killing of HCC cells by inhibition of Bcl-2 activity 43 and death receptor–mediated apoptosis is regulated by Bcl-2 pro- and anti-apoptotic protein balance. 44 Moreover, the siRNA-mediated Bcl-xL depletion experiments provided further evidence to show that Bcl-xL activity is critical for protecting cells against Lexa- or PF-induced apoptosis (Figure 5). Overall, these data suggest that Bcl-xL over-expression may be not a rare event and may contribute to pancreatic cancer development and resistance to chemotherapy. Our findings also imply that pancreatic tumors may be susceptible to treatment by Bcl-xL activity inhibition.
The anticancer effects of this combination with PF and Lexa via inhibiting Bcl-xL and activate DR5 are further confirmed in a mouse model burden with pancreatic cancer. Additionally, we observed that Caspase 3 was activated in samples synergistically administrated with Lexa and PF, suggesting that Lexa and PF co-treatment could induce apoptosis in vivo. Taken together, our findings imply that blockage of oncogene protein Bcl-xL is indeed crucial for targeting apoptosis induced by chemotherapeutics with either Lexa or PF. This could be developed to be used as part of a novel chemotherapy treatment in combination with surgical resection to improve patient survival in the future.
Footnotes
Acknowledgements
The authors would like to thank Dr Robin Humphreys and Dr Mani Subramanian of Human Genome Science for providing Lexatumumab.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical statement
With the approval by the University of Florida Gainesville Health Science Center Institutional Review Board (IRB-01) and with informed written consent, pancreatic cancer tissues and non-tumor liver tissues from same patients, respectively, were collected and analyzed.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported, in part, by a National Institutes of Health grant (CA133086 and RR023976) to C.L., National Natural Science Foundation of China (31371425) to X.Z., and Liaoning Provincial Natural Science Foundation of China (2013023056) to X.Z.