
Abstract
hMLH1 is one of the mismatch genes closely related to the occurrence of gastric cancer. Epigenetic regulation may play more important roles than gene mutations in DNA damage repair genes to drive carcinogenesis. In this article, we discuss the role of epigenetic changes, especially histone modifications in the regulation of hMLH1 alternative splicing. Our results showed that hMLH1 delEx10, delEx11, delEx10–11, delEx16 and delEx17 transcripts were ubiquitous in sporadic Chinese gastric cancer patients and gastric cancer cell lines. Lower level of H4K16ac and H3ac was detected in hMLH1 exon 10–11 region in gastric cancer cell lines when compared with human gastric mucosal epithelial cell line GES-1. A significant decrease of hMLH1 delEx11 and delEx10–11 was observed in gastric cancer cell lines after trichostatin A treatment. H3K36me3 and H3K4me2 levels were lower in hMLH1 exon 10–11 and exon 16–17 regions in gastric cancer lines when compared with GES-1. Aberrant transcripts such as hMLH1 delEx11 and delEx10–11 were significantly higher in gastric cancer cell lines after small interfering RNA–mediated knockdown of SETD2 (the specific methyltransferase of H3K36). The hMLH1 delEx10 and delEx10–11 transcripts were increased after interference of SRSF2. Taken together, our study demonstrates that lower level of histone acetylation and specific histone methylation such as H3K36me3 correlate with aberrant transcripts in hMLH1 exon 10–11 region. SRSF2 may be involved in these specific exons skipping as well.
Introduction
Gastric cancer (GC) is the fifth most common malignancy and the third leading cause of cancer death worldwide, with high incidence rate and poor prognosis in Eastern Asia, especially in China. 1 Gastric carcinogenesis is a complex and multifactorial process, in which the accumulation of multiple genetic and epigenetic changes plays a major role. 2
hMLH1 (human mutL homolog 1) is one of the DNA mismatch repair (MMR) genes closely related to the occurrence of GC, which is located in chromosome 3p22.3, consisting of 19 exons. 3 Inactivation of hMLH1 was found to be responsible for the development of GC exhibiting high-frequency microsatellite instability (MSI). 4 In our previous work, we identified several germline mutations of hMLH1 in patients with gastrointestinal cancer. 5 Despite the finding of genetic mutations, the majority of expressional and translational diversity of hMLH1 protein due to aberrant splicing of hMLH1 in GC cannot be explained.
Alternative splicing is a crucial step of the gene expression process in eukaryotes.6–8 It is a major cause for protein diversity and plays critical roles in cell development and tissue differentiation.9–11 Recent groundbreaking studies have disclosed that epigenetic factors, including DNA methylation, chromatin structure, and histone modifications, interact with each other and regulate the process of alternative pre–messenger RNA (mRNA) splicing, forming a large and complex regulatory network. 12 Besides, long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) have also been involved in the mediation of mRNA stability and splicing via combining with histone-modifying complexes and then targeting on DNA.13–15
In this study, we screened for aberrant splicing transcripts of hMLH1 in GC patients and discussed the role of DNA methylation and histone modifications in the regulation of hMLH1 pre-mRNA alternative splicing. By analyzing the relationship between the occurrence of aberrant splicing and epigenetic modifications in surrounding areas, we hope to probe the regulatory mechanism of hMLH1 alternative splicing.
Materials and methods
Individuals and samples
In total, 30 GC patients from the East District of China who have been histologically diagnosed between 1 January 2009 and 30 June 2012 were included in this study. Both the tumor tissues and adjacent tissues were collected. Detailed information is summarized in Table 1, including age of onset, gender, GC family history, and TNM staging classifications. All participants in the study provided written informed consent, and this study was approved by the Ethics Committee of the Medical School of Nanjing University.
The clinical data of 30 GC patients.

GC: gastric cancer.
Individuals with GC and one or more first-degree relative with GC or related cancers.
According to TNM Staging Classification for Carcinoma of the Stomach in NCCN guidelines (7th ed., 2010) by the American Joint Committee on Cancer (AJCC).
Cell culture
The GC cell lines SGC-7901, BGC-823, and MGC80-3 were poorly differentiated adenocarcinoma cells purchased from Shanghai Cell Bank of Chinese Academy of Sciences, China. Human gastric mucosal epithelial cell line GES-1 was purchased from Cell bank of Xiangya Medical School, Central South University, China. All these cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum at 37°C with 5% CO2. Emetine was added to100 µg/mL for 8 h before harvest of the cells.
Bioinformatics analysis
NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html) was used to predict potential acceptor sites and donor sites in hMLH1. 16
RNA extraction and reverse transcription polymerase chain reaction
Total RNA from frozen GC tissues, the paired adjacent normal tissue, GC cell lines, and GES-1 cell line was extracted using RNAiso Plus (TaKaRa Biotechnology (Dalian) Co., Ltd.). Reverse transcription polymerase chain reaction (RT-PCR) was performed in two steps. First-strand complementary DNA (cDNA) synthesis was performed using PrimerScript RT reagent Kit (TaKaRa) with random DNA hexamers and oligo-dT primer according to the manufacturer’s protocol. cDNA was amplified in the region of exons 9–12 and exons 15–19 of hMLH1. Primer sequences are as follows: hMLH1-Ex10–11-F: 5′-TGGAAATGCTGTTAGTCGAGAA-3′, hMLH1-Ex10–11-R: 5′-AGACGAGGTCAGACTTGTTGTG-3′, hMLH1-Ex16–17-F: 5′-CACCAAGCTTAGTGAAGAACTG-3′, and hMLH1-Ex16–17-R: 5′-GAAAGAAGAACACATCCCAC-3′. PCR conditions were as follows: an initial reaction at 95°C for 5 min; 40 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s; and followed by a final extension at 72°C for 5 min. Agarose gel electrophoresis was carried out using 1.5% gels run at 100 V for 40 min. The purified amplification products were clone sequenced on the ABI 3130-Avant automated sequencer (Applied Biosystems, Foster City, CA, USA).
DNA methylation assay
DNA was treated with bisulfite using the CpGenome™ DNA Modification Kit (Chemicon International, Temecula, CA, USA) according to manufacturer’s protocol. Bisulfite treatment of genomic DNA could convert all unmethylated cytosine into uracil while keeping methylated cytosine unchanged. 17 Methylation status of CpG sites in hMLH1 exon 10–11 and exon 16–17 regions was analyzed by bisulfite sequencing PCR (BSP) on an ABI 3130-Avant automated sequencer (Applied Biosystems). Primer sequences for BSP are as follows: hMLH1-Ex10–11-BSP-F—5′-GATGTTAATGTGTATTTTATAAAGTATGAA-3′, hMLH1-Ex10–11-BSP-R—5′-AACAAACAAAAATCTAAACTCTCAC-3′, hMLH1-Ex16–17-BSP-F—5′-TTAGTTGGTTAATATGATGAAATTT-3′, and hMLH1-Ex16–17-BSP-R—5′-AAAAACCTAATCCTAACCTTAAAAC-3′.
Reverse transcription quantitative polymerase chain reaction
hMLH1 expression in GC specimens and cell lines is measured by SYBR Green real-time PCR on ABI StepOnePlus Real-Time PCR System (Applied Biosystems). The quantitative polymerase chain reaction (qPCR) mixture contained total RNA–derived cDNA about 100 ng, the forward primer, reverse primer, ROX Reference Dye, SYBR, and Premix Ex Taq™ (TaKaRa Biotechnology (Dalian) Co., Ltd.). The thermal cycle conditions for assay were as follows: 95°C at 30 s, 40 cycles at 95°C for 15 s, and 60°C for 30 s. The 2−ΔCT method of relative quantification was calculated to determine the fold change in expression. The expression of actin was used as reference mRNA. Each RT-qPCR assay was done in triplicate. Sequences of PCR primers are listed in Supplementary Table 1.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were done by using the EZ-Magna ChIP Kit (catalog no. 17-408; Millipore, billerica, MA, USA) under the instruction of manufacturer’s protocol. The detailed procedure is the same as previously reported. 18 Primers are described in Supplementary Table 2. The qPCR reaction condition is the same as mentioned above.
In RNA splicing analysis, the GC cell lines SGC-7901, BGC-823, and MGC80-3 and human gastric mucosal epithelial cell line GES-1 were treated with DNA methyltransferase inhibitor 5-aza 2′-deoxycytidine (AZA) or histone deacetylase (HDAC) inhibitor trichostatin A (TSA) in order to analyze whether hMLH1 alternative splicing is affected by DNA methylation or histone acetylation status. During cell culture, 1 µM of 5-aza 2′-deoxycytidine (AZA; Sigma–Aldrich, St. Louis, MO, USA), a kind of DNA methyltransferase inhibitor, was added to the medium and maintained for 72 h. A volume of 0.5 µM of TSA (Sigma-Aldrich) was added and maintained for 12 h. hMLH1 RNA expression condition was measured by RT-qPCR.
siRNA-mediated knockdown of SERD2 or SRSF2
Downregulation of SETD2 or SRSF2 was achieved by using siRNA oligonucleotides (RiboBio Co., Ltd., Guangzhou, China) to interfere its expression. siRNA oligonucleotides against human SETD2 or SRSF2 were added to cells at 50 nM according to the manufacturer’s instructions. Cells were harvested and the interference efficiencies were analyzed by RT-qPCR 48 h after transfections.
Statistical analysis
Relationship between clinical pathological data and aberrant transcript levels of GC patients was analyzed by two-sided χ2 test or Fisher’s exact test. Analysis of variance (ANOVA) and Student–Newman–Keuls (SNK)-q test were used to analyze the differences in transcript levels between different cell lines and treatment groups. All statistical tests were two-sided and were performed by the SPSS software package (version 16). The p < 0.05 was considered statistically significant.
Results
Bioinformatics analysis of hMLH1 sequence
As shown in Figure 1, two acceptor sites were found in hMLH1 exon 10 and exon 11 by NNSPLICE suggesting that aberrant transcripts such as loss of exon 10 or 11 of hMLH1 (hMLH1 delEx10, delEx11, and delEx10–11) may be produced if the weak acceptor site is identified by the spliceosome. There also exist extremely weak donor sites in hMLH1 exon 16 and weak acceptor sites in exon 17 indicating the possibility of loss of exon 16 or 17 of hMLH1 (hMLH1 delEx16 and delEx17).

Splice sites prediction by bioinformatic analysis. Capital letters show hMLH1 exon sequence and lowercases represent flanking intron sequences. (a) NNSPLICE predicted that there are two acceptor sites (acceptor site 1, tgctttctttttattgtttagATCGTCTGGTAGAATCAACT, score 0.99; acceptor site 2, ACACCCATTCCTGTACCTCAGgtaatgtagcaccaaactcc, score 0.88) and one donor site (ACCTCAGgtaatgta, score 0.93) in hMLH1 exon 10 and flanking sequences. (b) NNSPLICE predicted that there are two acceptor sites (acceptor site 1, tctctcttattttcctgacagTTTAGAAATCAGTCCCCAGA, score 0.94; acceptor site 2, CCTG GGCTCCAATTCCTCCAGGATGTACTTCACCCAGgtca, score 0.66) and one donor site (CACCCAGgtcagggc, score 0.86) in hMLH1 exon 11 and flanking sequences. (c) NNSPLICE predicted that there are two acceptor sites (acceptor site 1, catgttcttgcttcttcctagGAGCCAGCACCGCTCTTTGA, score 0.99; acceptor site 2, ACCTTGCCATGCTTGCCTTAGATAGTCCAGAGAGTGGCTGG, score 0.71) and one extremely weak donor site (TGATGAGgtgtgaca, score 0.31) in hMLH1 exon 16 and flanking sequences. (d) NNSPLICE predicted that there are one extremely weak acceptor site (ccttgtccttt ttcctgcaagcagGAAGGGAACCTGATTGG, score 0.43) and one donor site (CACTGAGgtcagtga, score 0.91) in hMLH1 exon 17 and flanking sequences.
hMLH1 delEx10, delEx11, delEx10–11, delEx16, and delEx17 transcripts were found in Chinese GC patients
According to the bioinformatics analysis of hMLH1 sequence, we designed primers targeting relevant areas. RT-PCR and clone sequencing revealed the coexistence of hMLH1 wild-type transcript and several distinct shorter transcripts hMLH1 delEx10, delEx11, delEx10–11, delEx16, and delEx17 in GC patients’ tumor tissues (Figure 2). The delEx10, delEx11, and delEx10–11 transcripts were predicted to change the reading frame, create a premature termination codon (PTC), and then produce nonfunctional truncated proteins of 265 aminos, 315 aminos, and 292 aminos, respectively (Figure 2(a)). The delEx16 and delEx17 transcripts will lead to in-frame deletion and cause the loss of 55 or 31 amino acids in the carboxyl terminal of hMLH1 protein (Figure 2(b)).
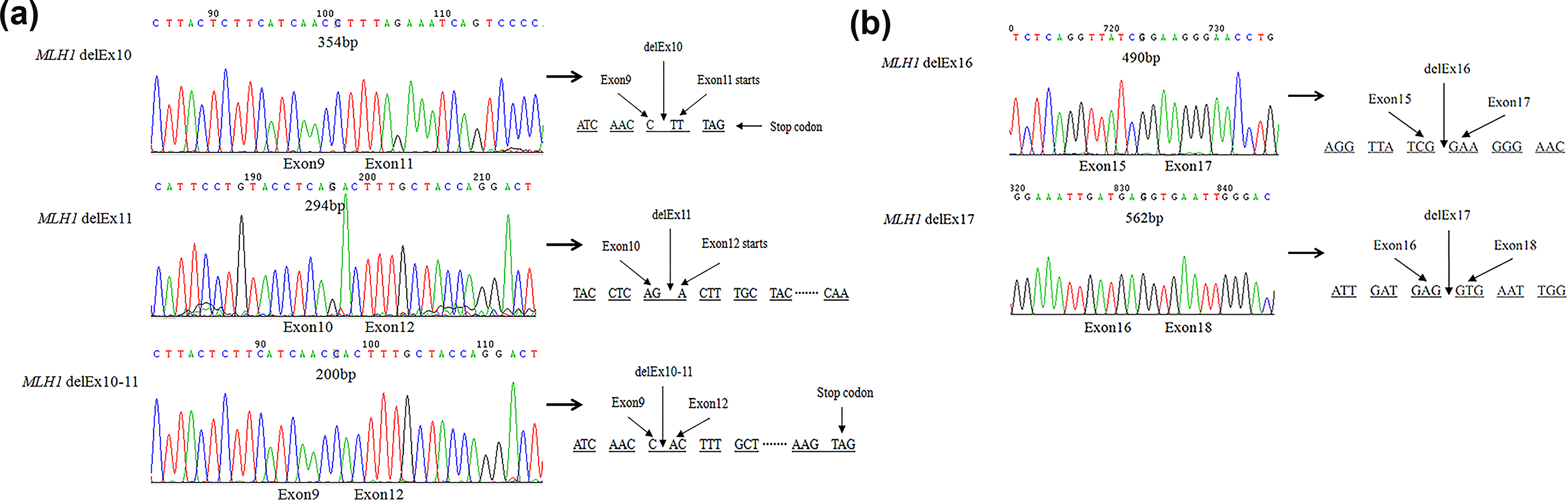
RNA splicing analysis of different transcripts in hMLH1 exon 10–11 and exon 16–17 regions in GC patients. (a) Cloning sequencing of RT-PCR products from tumor tissue in GC patients with primers targeting hMLH1 exon 10–11 region. Sequencing of 354bp, 294bp, and 200bp PCR products revealed a transcript demonstrating loss of exons 10, 11, and 10–11, respectively. The delEx10, delEx11, and delEx10–11 transcripts were predicted to change the reading frame, create a premature termination codon (PTC), and then produce nonfunctional truncated proteins of 265 aminos, 315 aminos, and 292 aminos. (b) Cloning sequencing of RT-PCR products from tumor tissues in GC patients with primers targeting hMLH1 exon 16–17 region. Sequencing of the 490bp and 562bp PCR products demonstrated the existence of delEx16 and delEx17 transcripts as predicted by bioinformatics analysis. The delEx16 and delEx17 transcripts will lead to in-frame deletion and cause the loss of 55 or 31 amino acids in the carboxyl terminal of hMLH1 protein.
To determine whether these aberrant splicing transcripts exist widely in GC patients, RT-qPCR was conducted on tumor and paired normal tissues from 30 GC patients with no relevant germline mutation. Results suggest that normal transcripts were lower in tumor tissue than in the paired normal tissue in half of these patients (Figure 3(a) and (d)). Besides, the delEx10, delEx10–11, delEx16, and delEx17 transcripts were detected in most of these patients both in the tumor tissues and the paired normal tissues (Figure 3). It must be noted that this result might be an underestimation of these aberrant transcripts since the nonsense-mediated decay (NMD) pathway should be taken into consideration. Triggered by the premature terminal codon, NMD might result in constant rapid degradation of aberrant transcripts, thus increase the ratio of normal transcripts to some extent. Due to the limited size of samples and effect of NMD pathway, delEx11 transcripts were not detected. No significant difference was found in the relationship between the aberrant splicing transcripts and clinical pathological data of GC patients, indicating that the occurrence of alternative splicing in GC patients was not affected by age, gender, family history, and TNM staging (Table 2).
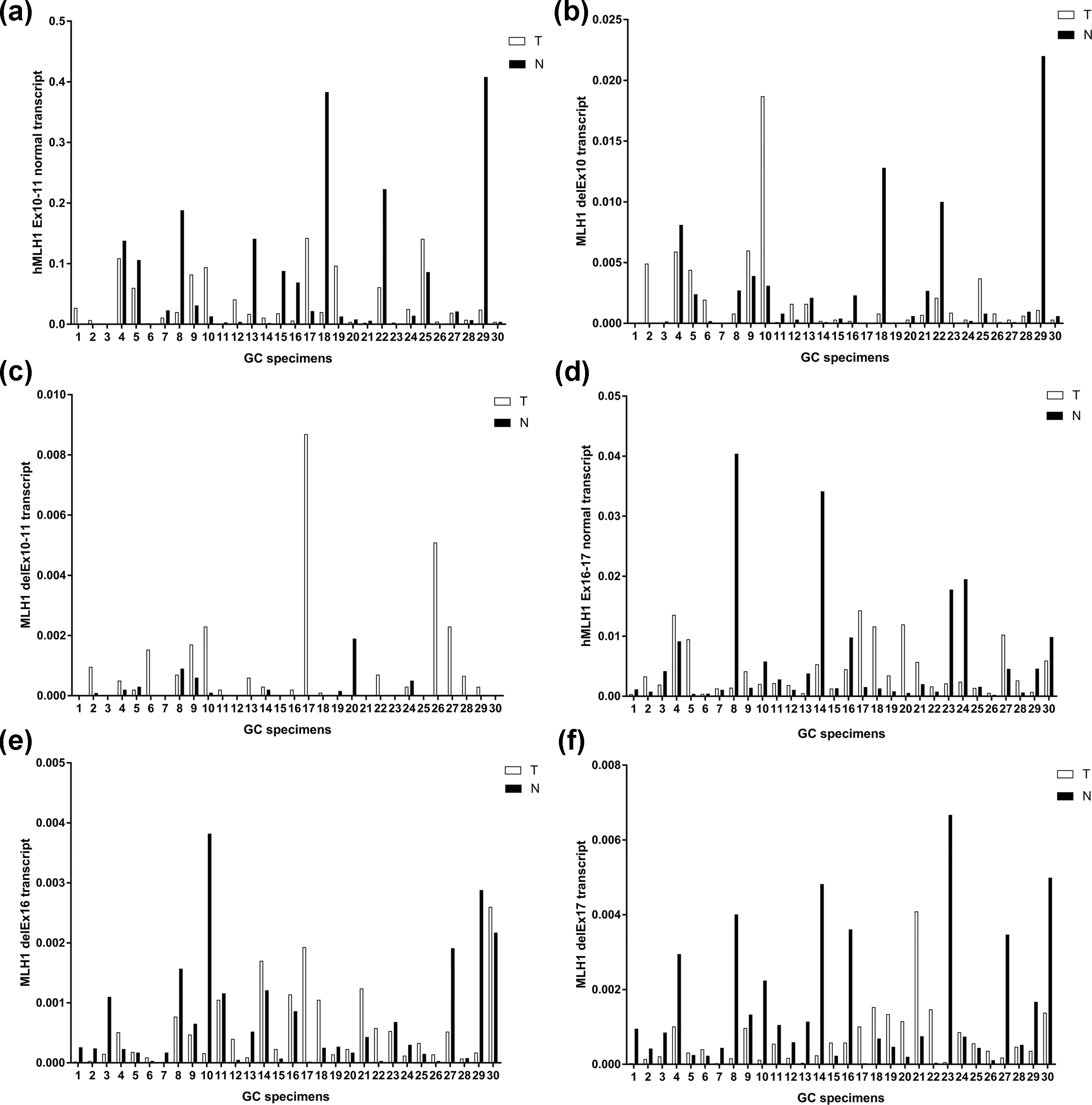
Expression level of hMLH1 in 30 matched GC specimens. The bar diagram is an expression of hMLH1 in GC specimens (white) and paired normal specimens (black) adjusted to actin. T: tumor specimens and N: paired normal specimens. Expression level of (a) hMLH1 Ex10–11 normal transcript, (b) hMLH1 delEx10 transcript, (c) hMLH1 delEx10–11 transcript, (d) hMLH1 Ex16–17 normal transcript, (e) hMLH1 delEx16 transcript, and (f) hMLH1 delEx17 transcript in tumor tissues and paired normal tissues.
The relationship between occurrence of aberrant splicing and clinical pathological data in 30 GC patients.

GC: gastric cancer.
Two-sided χ2 test or Fisher’s exact test.
Individuals with GC and one or more first-degree relative with GC or related cancers.
According to TNM Staging Classification for Carcinoma of the Stomach in NCCN guidelines (7th ed., 2010) by the American Joint Committee on Cancer (AJCC).
hMLH1 delEx10, delEx11, delEx10–11, delEx16, and delEx17 transcripts were more common in GC cell lines than human gastric mucosal epithelial cell line GES-1
RT-qPCR was performed to compare the levels of hMLH1 transcripts between GC cell lines and GES-1 cell line. A lower expression of normal hMLH1 transcript was detected in GC cell lines SGC-7901, MGC80-3, and BGC-823 compared to GES-1 (Figure 4(a)). A trend of higher ratio of hMLH1 delEx10, delEx11, and delEx10–11 transcripts versus hMLH1 normal transcript was found in GC cell lines when compared with GES-1 cells (Figure 4(b)–(d)), except for the lower expression level of hMLH1 delEx10–11 transcript in MGC80-3 than that of GES-1 cells probably due to the higher proportion of hMLH1 delEx11 in MGC80-3 (Figure 4(d)). The ratio of hMLH1 delEx16 versus hMLH1 normal transcript is significantly higher in MGC80-3 than GES-1 (Figure 4(e)). A higher proportion of hMLH1 delEx17 versus hMLH1 normal transcript is found in BGC-823 compared with GES-1 (Figure 4(f)). Though the proportion of these aberrant splicing transcripts is not the same in different GC cell lines, there is a tendency that the expression levels of alternative splicing transcripts are generally higher in GC cell lines than those of GES-1, indicating that the increasing ratio of aberrant splicing may be closely related to the occurrence of GC.

Expression level of hMLH1 transcripts in GC cell lines SGC-7901, BGC-823, and MGC80-3 and human gastric mucosal epithelial cell line GES-1. (a) Expression level of hMLH1 normal transcript in GC cell lines and GES-1. (b)–(f) Ratios of hMLH1 aberrant transcripts versus hMLH1 normal transcript in GC cell lines and GES-1. The expression level in GES-1 was set to 1 (*p < 0.05, indicating statistical significance).
DNA methylation may not affect the amount of hMLH1 full length and aberrant splicing transcripts
Hypermethylation was observed at the CpG sites of hMLH1 Ex10–11 and hMLH1 Ex16–17 in GC cell lines and human gastric mucosal epithelial cell line GES-1 by BSP (Figure 5). No significant difference was found in the methylation level of relevant region in hMLH1 among these four cell lines. To further explore the effect of DNA methylation on alternative splicing, we treated the three GC cell lines SGC-7901, BGC-823, and MGC80-3 and human gastric mucosal epithelial cell line GES-1 with DNA methyltransferase inhibitor AZA. As shown in Figure 6, AZA treatment did not change ratios of hMLH1 delEx10, delEx11, delEx10–11, delEx16, and delEx17 transcripts versus hMLH1 normal transcript in all these four cell lines suggesting that DNA methylation may not affect the amount of these aberrant transcripts in GC cell lines.
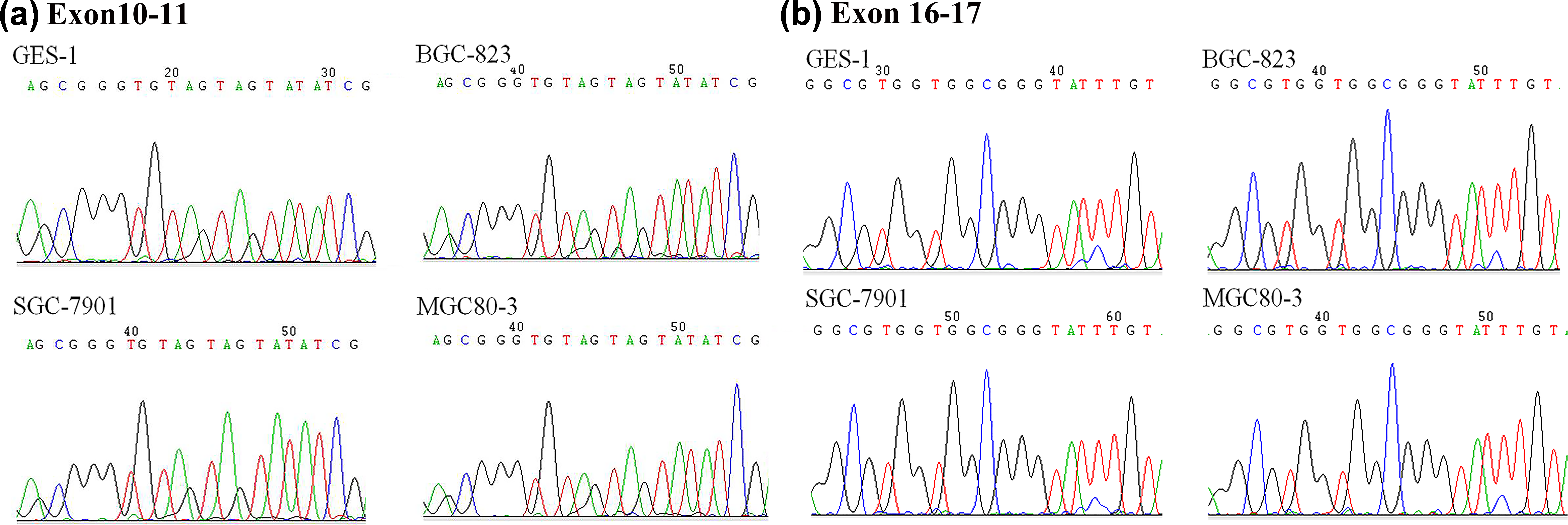
Methylation status of (a) hMLH1 exon 10–11 region and (b) hMLH1 exon 16–17 region in human gastric mucosal epithelial cell line GES-1 and the GC cell lines SGC-7901, BGC-823, and MGC80-3. DNA isolated from cells shows a high C content at all CpGs because of hypermethylation of the DNA.
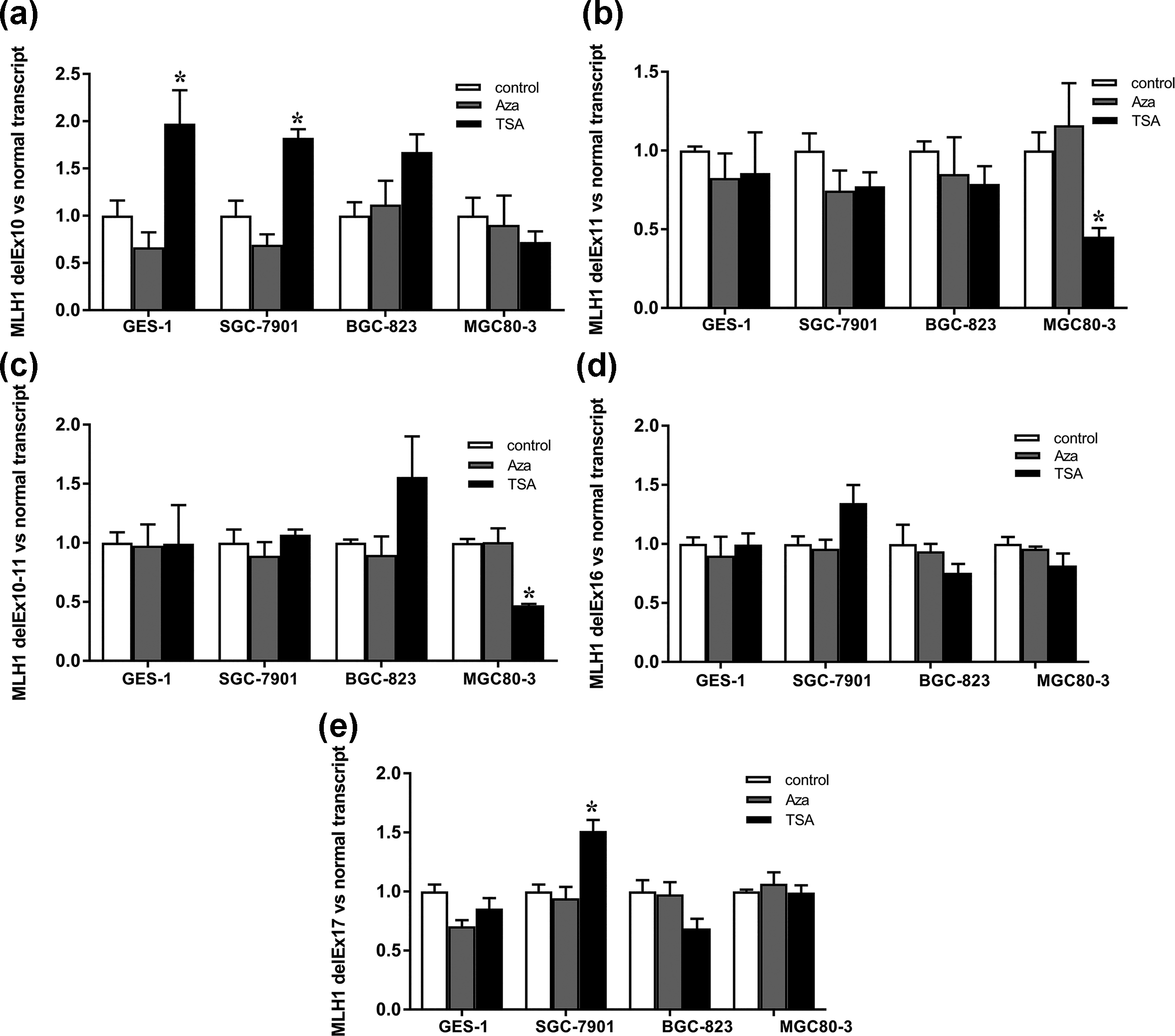
The relative folds of ratios of hMLH1 aberrant transcripts versus hMLH1 normal transcript in GC cell lines SGC-7901, BGC-823, and MGC80-3 and human gastric mucosal epithelial cell line GES-1 after AZA or TSA treatment. (a) hMLH1 delEx10 versus hMLH1 normal transcript. (b) hMLH1 delEx11 versus hMLH1 normal transcript. (c) hMLH1 delEx10–11 versus hMLH1 normal transcript. (d) hMLH1 delEx16 versus hMLH1 normal transcript. (e) hMLH1 delEx17 versus hMLH1 normal transcript. The values were calculated as (2−ΔCt (aberrant transcript-actin)/2−ΔCt (normal transcript-actin)). Relative folds of ratios of untreated cell lines serve as 1 (*p < 0.05, indicating statistical significance).
Low level of histone acetylation was detected in hMLH1 exon 10 and exon 11 regions. Histone acetylation may not be involved in the regulation of hMLH1 exon 16–17 alternative splicing
ChIP was performed to get the whole picture of the distribution of histone modifications along whole sequence of hMLH1. Lower level of H4K16ac and H3ac was detected in hMLH1 exon 10 and exon 11 regions in SGC-7901 and BGC-823 when compared with GES-1 (Figure 7). After promoting histone acetylation with HDAC inhibitor TSA, the downward ratios of hMLH1 delEx11 and delEx10–11 transcripts versus hMLH1 normal transcript were observed in MGC80-3 (Figure 6(b) and (c)). While in hMLH1 exon 16–17 region, there is no apparent difference in the enrichment of H4K16ac and H3ac in SGC-7901 and BGC-823 versus GES-1 (Figure 7). No regular changes in hMLH1 delEx16 and delEx17 transcripts were found after TSA treatment (Figure 6(d) and (e)). These results suggest that histone acetylation may affect the outcome of alternative splicing in hMLH1 exon 10–11 region. A more compact chromatin structure owing to the lower level of histone acetylation may slow the elongation rate of RNA polymerase II (RNA Pol II) and then favor the identification of weak splice sites resulting in exon exclusion. This effect was counteracted by treating the cells with TSA, which served as a chromatin “opener,” allowing for the rapid elongation of RNA Pol II so that only strong splice sites can be identified by spliceosome.

The abundance of H3K36me3, H3K4me2, H4K16ac, and H3ac in different regions of hMLH1 in (a) SGC-7901 and (b) BGC-823 when compared with GES-1.
Low level of H3K36 tri-methylation in hMLH1 exon 10–11 region was correlated with relevant exon skipping
To analyze whether the exon skipping could be modulated by histone methylation patterns, we performed ChIP assay using antibodies recognizing methylation at different lysines of histone H3 to evaluate the accumulation level of histone methylation in hMLH1 whole sequence. H3K36me3 and H3K4me2 levels were lower in hMLH1 exon 10–11 and exon 16–17 regions in SGC-7901 and BGC-823 when compared with GES-1 (Figure 7). To further confirm the effect of H3K36me3 on alternative splicing patterns, modulation of H3K36me3 level was done by downregulation of its specific histone methyltransferases SETD2. A value of more than 80% SETD2 knockdown was determined by qPCR analysis after siSETD2-003 treatment of SGC-7901, BGC-823, and GES-1 (Figure S1). The ratios of hMLH1 delEx11 and delEx10–11 transcripts versus normal transcript were significantly higher in BGC-823 transfected with SETD2 siRNA compared to cells transfected with scramble (non-targeting) siRNA (Figure 8(a)–(c)). These results revealed a strong correlation between H3K36me3 across the alternatively spliced regions of hMLH1 exon 10–11 and splicing outcome. Low level of H3K36me3 along the alternatively spliced regions may lead to poor recruitment of splicing factors to the normal acceptor site of nascent RNA as a consequence favoring accumulation of aberrant transcripts. siSETD2 treatment did not influence ratios of hMLH1 delEx16 and delEx17 transcripts versus normal transcript in all three kinds of cells, suggesting that H3K36me3 may be not involved in the regulation of alternative splicing in hMLH1 exon 16–17 (Figure 8(d) and (e)).

The relative folds of ratios of hMLH1 aberrant transcripts versus hMLH1 normal transcript in GES-1, SGC-7901, and BGC-823 after siSETD2-003 treatment. (a) hMLH1 delEx10 versus hMLH1 normal transcript. (b) hMLH1 delEx11 versus hMLH1 normal transcript. (c) hMLH1 delEx10–11 versus hMLH1 normal transcript. (d) hMLH1 delEx16 versus hMLH1 normal transcript. (e) hMLH1 delEx17 versus hMLH1 normal transcript. The values were calculated as (2−ΔCt (aberrant transcript-actin)/2−ΔCt (normal transcript-actin)). Relative folds of ratios of untreated cell lines serve as 1 (*p < 0.05, indicating statistical significance).
Knockdown of SRSF2 influences hMLH1 alternative splicing patterns in exon 10–11 region
SRSF2 is a kind of splicing factor targeting exonic splicing enhancer which promotes the formation of spliceosome. Si-RNA-specific targeting SRSF2 was used to silence the expression of SRSF2. The interference efficiencies were shown to be more than 80% tested by qPCR (Figure S2). After siSRSF2-001 treatment of SGC-7901, BGC-823, and GES-1, the ratio of hMLH1 delEx10 and delEx10–11 transcripts versus normal transcript increased in all three cell lines, indicating that SRSF2 may favor inclusion of exon by recruiting splicing factors to normal splice sites (Figure 9(a) and (c)). No change in aberrant transcript levels in hMLH1 exon 16–17 region was observed after interference of SRSF2 (Figure 9(d) and (e)).

The relative folds of ratios of hMLH1 aberrant transcripts versus hMLH1 normal transcript in GES-1, SGC-7901, and BGC-823 after siSRSF2-001 treatment. (a) hMLH1 delEx10 versus hMLH1 normal transcript. (b) hMLH1 delEx11 versus hMLH1 normal transcript. (c) hMLH1 delEx10–11 versus hMLH1 normal transcript. (d) hMLH1 delEx16 versus hMLH1 normal transcript. (e) hMLH1 delEx17 versus hMLH1 normal transcript. The values were calculated as (2−ΔCt (aberrant transcript-actin)/2−ΔCt (normal transcript-actin)). Relative folds of ratios of untreated cell lines serve as 1 (*p < 0.05, indicating statistical significance).
Discussion
The hMLH1 protein, composed of 756 amino acids, is a MMR enzyme recruiting other DNA-repair proteins to the MMR complex. 19 A series of studies have demonstrated that germline mutation of hMLH1 and DNA methylation of the hMLH1 promoter are strongly associated with a loss of expression of the gene, resulting in MSI in GC.20,21 However, in our previous work, we found a certain proportion of GC patients who lack hMLH1 expression with no germline mutation of hMLH1, nor did the hypermethylation of promotor being detected, which prompt us to further explore other mechanisms of the hMLH1 gene inactivation in GC cells. 5
In our study, we used NNSPLICE to analyze the whole sequence of hMLH1, predicting potential alternative splicing patterns according to the strength of splice sites. NNSPLICE predicted that there are two acceptor sites and one donor site in hMLH1 exon 10 or exon 11 and flanking sequences. Generally, normal transcripts will be produced when the strong acceptor site is identified by spliceosome. In some circumstances such as the structural change of splicing-related RNA motif or changes in epigenetic modification may favor the identification of cryptic acceptor site and thus increase the accumulation of hMLH1 delEx10, delEx11, and delEx10–11 transcripts which may produce nonfunctional truncated proteins. Additionally, in hMLH1 exon 16–17 region, the scores of donor site in exon 16 and acceptor site in exon 17 are extremely lower than average, making exon exclusion much more easier.
To determine whether these aberrant splicing transcripts really exist in GC, RT-PCR was conducted on tumor tissues and paired normal tissues in GC patients. The existence of delEx10, delEx11, delEx10–11, delEx16, and delEx17 transcripts was confirmed in most of the GC patients’ tumor tissues and the paired normal tissues, indicating that these alternative splicing patterns are universal phenomena. We further show that higher level of aberrant transcripts was observed in GC cell lines versus human gastric mucosal epithelial cell line GES-1. There might be a natural balance between normal and abnormal alternative splice products. 22 Once the delicate balance ever fails, functional defects of hMLH1 protein may lead to genetic instability which in turn increases the tumor susceptibility. 23
The existence of hMLH1 delEx16, delEx17, and delEx10–11 transcripts and their potential relevance to diseases has been reported in previous studies.24–27 However, hMLH1 delEx10 or delEx11 transcripts have not been reported. The cause of occurrence and pathogenic role of all these aberrant hMLH1 transcripts in GC have not been extensively assessed either. Why these aberrant transcripts own a high frequency in GC? What is the potential regulatory mechanism that promotes these exons skipping? It has been realized that a full understanding of alternative splicing regulation not only requires analysis of RNA sequence elements and their associated splicing factors but also relies on epigenetic components such as chromatin structure, DNA methylation, and histone modification.11,12,28 According to our previous screening of hMLH1 mutation, mutation was not detected in exon 10–11, exon 16–17, and the corresponding intron sequence in those selected GC patients suggesting that the influence of genetic mutation on alternative splicing can be ruled out.5,29 Hence, we propose a hypothesis that epigenetic modifications might play an important role in the regulation of hMLH1 alternative splicing.
Recent provocative studies point out that DNA methylation may affect splice site choice directly through coupling with adaptor protein or affect splicing outcome via changes in histone modifications.30–33 From our data, there is no obvious correlation between DNA methylation and the occurrence of these aberrant splicing. Changes in methylation status by AZA do not affect the balance of alternative splicing. DNA methylation might be critical in some genes while not in hMLH1. 32
Given the realization that histone modifications can affect alternative splicing, it is attractive to speculate that a histone-based system may encode information in the selection of alternative splicing patterns in cell types and tissues.34–36 In our study, a significant decrease in acetylation for histones H3 and H4K16Ac surrounding exon 10–11 region was detected in GC cell lines. An additional indication for a role of histone acetylation in alternative splicing is the observation that treatment with the HDAC inhibitor TSA induces downregulation of hMLH1 delEx11 and delEx10–11 transcripts in MGC80-3 cells. This reversible effect may be due to an intragenic modulation of the RNA Pol II elongation rate. 37 As shown in Figure 1(a) and (b), there are two alternative acceptor sites in hMLH1 exon 10 or exon 11 and flanking sequences (one strong acceptor site and one weak cryptic acceptor site). Lack of histone acetylation in GC cells favors the formation of a more compact chromatin structure, thus slowing down transcriptional elongation rate of RNA Pol II resulting in the recognition of weaker splice site so that the accumulation of aberrant splicing transcripts is enhanced. Treatment with potent inhibitor of histone deacetylation will increase acetylation of H3 and H4 allowing for the rapid elongation of RNA Pol II which favors the recruitment of the splicing machinery to strong splice site.38,39 However, there is no apparent difference between GC cell lines and GES-1 in the abundance of H4K16ac and H3ac in hMLH1 exon 16–17 region. No noticeable changes in the expression of hMLH1 delEx16 and delEx17 transcripts in GC cell lines ever being observed after TSA treatment.
Histones such as H3K36me3 and H3K4me2 are also typical histone markers which are differentially distributed along genes. 40 Several lines of evidence show that they could affect the outcome of alternative splicing either through modulation of RNA Pol II elongation rate or through specific recruitment of splicing factors via adaptor complexes to weak RNA-binding sites.36,37,41–43 In our study, analysis of histone methylation revealed a lower level of H3K36me3 surrounding hMLH1 exon 10–11 and exon 16–17 in SGC-7901 and BGC-823 when compared with GES-1. Modulation of H3K36me3 level by knockdown of the specific histone methyltransferases SETD2 shows a shift of alternative splicing pattern in favor of hMLH1 delEx11 and delEx10–11 transcripts in BGC-823. These observations point to a role for histone methylation especially H3K36me3 in the regulation of alternative splicing in hMLH1 exon 10–11. Previous study suggests that the abundance of H3K36me3 may directly or indirectly modulate RNA Pol II density at exons and therefore affect splicing efficiency.43,44 Luco found an additional mechanism that H3K36me3 can create a platform on chromatin for the recruitment of polypyrimidine tract–binding protein (PTB) to the nascent RNA via an adaptor protein, MRG15. The richness of H3K36me3 along the alternatively spliced region of the gene may attract MRG15 which interacts with PTB and thus recruit it to the nascent RNA causing the exclusion of PTB-dependent exon. 36 In line with the theory of chromatin-splicing adaptor system, we propose that the H3K36me3 mark might be recognized by a certain kind of chromatin binding proteins, which directly recruits splicing factors such as SRSF2 to the exonic splicing enhancer element surrounding alternative splice sites as a consequence favoring the inclusion of hMLH1 Ex10 and Ex11. This hypothesis is partially verified by the effect of SRSF2 on hMLH1 alternative splicing showing that the ratios of hMLH1 delEx10 and delEx10–11 transcripts versus normal transcripts in GC cell lines were increased after interference of SRSF2 (Figure 9(a) and (c)). Directed experiments to test the change in alternative splicing patterns after overexpression of H3K36me3 in the absence of SRSF2 are required in order to further verify the hypothesis that interaction of H3K36me3 with SRSF2 is critical in the regulation of hMLH1 alternative splicing. Further study to find the chromatin binding protein which links H3K36me3 and SRSF2 in hMLH1 exon 10–11 splicing is also needed. The concrete regulatory effect of H3K36me3 on alternative splicing is still flexible and varied in different genes and genes’ region. In spite of a lower level of H3K36me3 along hMLH1 exon 16–17 region in GC cell lines, it is still not sufficient to get a clear picture of the correlation between low level of H3K36me3 and exon 16–17 skipping since knockdown of SETD2 and SRSF2 did not influence ratios of hMLH1 delEx16 and delEx17 transcripts.
A hint toward a role for H3K4me3 in hMLH1 alternative splicing came from the significant lower level of H3K4me3 along hMLH1 exon 10–11 and exon 16–17. It has been reported that H3K4me3 favors the recruitment of the early spliceosome to human cyclin D1 pre-mRNA via interacting with the chromatin-adaptor protein CHD1. 41 However, the potential regulatory role H3K4me3 acts in this case seems not to be so simple. Additional study will be required to distinguish the effect of H3K4me3 on alternative splice site selection from indirect effects during transcription process.
Neither DNA methylation nor histone modification could explain the regulation of alternative splicing in hMLH1 exon 16–17 region. The selection of splice site in this region is far more complex than anticipated.
In summary, hMLH1 delEx10, delEx11, delEx10–11, delEx16, and delEx17 were ubiquitous in sporadic Chinese GC patients. There were more aberrant transcripts in GC cell lines than in normal gastric mucosal epithelial cell line GES-1. Low level of histone acetylation and histone methylation especially H3K36me3 along relevant regions correlate with exons’ exclusion in hMLH1 exon 10–11 region suggesting that a histone-based alternative splicing regulatory system might be involved in the selection of alternative splicing sites. However, the regulatory mechanism of exon 16 and exon 17 skipping is far more complicated and may be affected by crosstalk of different epigenetic modifications.
Footnotes
Acknowledgements
J.-X.Z. and X.-W.L. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by National Natural Science Foundation of China (No. 30972535) and Natural Science Foundation of Jiangsu, China (No. BK2012724).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.