
Abstract
A broad spectrum of tumors develop resistance to classic chemotherapy, necessitating the discovery of new therapies. One successful strategy exploits the synthetic lethality between poly(ADP-ribose) polymerase 1/2 proteins and DNA damage response genes, including BRCA1, a factor involved in homologous recombination–mediated DNA repair, and CDK12, a transcriptional kinase known to regulate the expression of DDR genes. CHK1 inhibitors have been shown to enhance the anti-cancer effect of DNA-damaging compounds. Since loss of BRCA1 increases replication stress and leads to DNA damage, we tested a hypothesis that CDK12- or BRCA1-depleted cells rely extensively on S-phase-related CHK1 functions for survival. The silencing of BRCA1 or CDK12 sensitized tumor cells to CHK1 inhibitors in vitro and in vivo. BRCA1 downregulation combined with CHK1 inhibition induced excessive amounts of DNA damage, resulting in an inability to complete the S-phase. Therefore, we suggest CHK1 inhibition as a strategy for targeting BRCA1- or CDK12-deficient tumors.
Introduction
A shared feature of various malignancies is the dysregulation of DNA damage response (DDR), which leads to genomic instability. 1 The checkpoint kinase 1 (CHK1) represents a cellular factor that could be used to target the viability of tumor cells with genomic instability.2–4 This is because CHK1 is involved in numerous essential cellular processes. For example, a cell responds to DNA damage by activating CHK1 through ATR-promoted phosphorylation, which effectively blocks cell cycle progression. Once activated, CHK1 regulates the G2/M checkpoint by inactivating the CDC25 phosphatases that would otherwise remove the inhibitory phosphates of cyclin-dependent kinases (CDK), which are responsible for the G2/M transition. 3 CHK1 also participates in the DNA damage repair mechanism by phosphorylating, and thus activating, the repair factors BRCA2 and RAD51. 5 In addition, CHK1 is integral to the prevention of replication stress, as it stabilizes replication forks and regulates origin firing. 3
Various CHK1 inhibitors have been tested as anti-tumor agents in combination with DNA-damaging agents, such as hydroxyurea, cisplatin, and topoisomerase inhibitors (topotecan, irinotecan), and antimetabolites, such as gemcitabine.6–10 Several studies have reported that the anti-tumor effect of CHK1 inhibitors is determined by p53 status, with p53-deficient cells more responsive to CHK1 inhibitor treatment.2,11 However, other authors have reported that CHK1 inhibitors decrease cellular viability irrespective of p53 suppressor status, 6 showing that CHK1 inhibitors strongly potentiate the effects of DNA-damaging agents in p53−/− cells. These results suggest that patients with p53-mutated tumors could benefit from treatment approaches that include CHK1 inhibition.11,12 The identification of cellular factors that, when combined with CHK1 inhibitors, confer synthetic lethality could strengthen the portfolio of combinatory treatments with CHK1 inhibitors or potentially lead to the discovery of monotherapy approaches for tumors with relevant mutations. Certain scenarios have been described recently; for example, cells deficient in Fanconi anemia genes are hypersensitive to CHK1 inhibition, and a novel essential interplay between CHK1 and I-kappa-B kinase epsilon was observed in ovarian cells.13,14
The CDK12 regulates the elongation phase of transcription by phosphorylating the C-terminal domain of RPB1, a subunit of RNA polymerase II (RNAPII).15–18 We previously identified CDK12 as a cellular factor that orchestrates the expression of several key DDR genes, for example, BRCA1, ATR, ATM, FANCI, and FANCD2. 16 By regulating DDR genes, CDK12 consequently affects homologous recombination (HR)-mediated DNA repair. A downregulation of CDK12 leads to increased endogenous DNA damage, DDR activation, and pronounced sensitivity to DNA-damaging agents.16,19 Cancer-associated CDK12 mutations are predominantly located within the kinase domain and result in a catalytically inactive protein. 20 Based on these observations, CDK12 has been suggested to be a tumor suppressor candidate.19–21
The breast cancer–associated gene 1 (BRCA1) tumor suppressor protein is a central component of several distinct protein complexes that are vital to HR-mediated DNA damage repair, cell cycle checkpoints, and transcriptional regulation.22,23 BRCA1 is inheritably mutated in about 9% and 13% of unselected women with newly diagnosed triple-negative breast cancer and ovarian cancer (respectively). If metastatic, these patients have generally very unfavorable prognosis and currently are candidates for targeted drug therapy, such as poly(ADP-ribose) polymerase (PARP) inhibitors.24–26 The loss of BRCA1, caused by homozygous mutations, reduces the ability of cells to carry out HR-mediated DNA repair, resulting in cellular genomic instability. 24 Interestingly, BRCA1 mutations are mutually exclusive with CDK12 mutations, which suggests that CDK12 belongs to the same HR-mediated DNA damage repair pathway as BRCA1. 21
HR deficiency presents an opportunity for cancer treatment. Tumors exhibiting HR deficiency, especially those with loss of BRCA1 or 2, are sensitive to inhibitors of PARP1/2, a protein involved in DNA repair. As with the loss of BRCA1/2, the loss or inhibition of CDK12 sensitizes cells to PARP inhibitors, which have recently been approved for the treatment of ovarian cancer.21,41 Nevertheless, research has reported that certain tumors have become resistant to PARP inhibitors as a result of restored HR capacity, altered non-homologous end-joining (NHEJ) capacity, decreased levels or activity of PARP1, and/or decreased intracellular availability of PARP inhibitors. 25 Thus, novel alternatives to PARP1 inhibitors are necessary for further patient treatment.
Although the genomic instability that results from DDR deficiency often drives tumor development, it also provides a great opportunity for cancer treatment. 26 The loss of BRCA1 and CDK12 function most likely potentiates the anti-tumor effects of PARP1/2 inhibitors by crippling HR-mediated DNA repair, with this mechanism a perfect example of the concept of synthetic lethality. Since the loss of BRCA1 compromises DDR and leads to replication stress and DNA damage, 27 we hypothesized that BRCA1- or CDK12-deficient cells will extensively rely on the S-phase-related kinase activity of CHK1 for survival. In this study, we demonstrate that silencing BRCA1 or CDK12 indeed sensitizes cancer cells to CHK1 inhibitors.
Materials and methods
Synthesis of CHK1 inhibitors
The racemic CHK1 inhibitor SCH900776 (Merck, Darmstadt, Germany) was prepared in-house through a previously published route.28,29 The enantiomers were separated by high-performance liquid chromatography (HPLC) with a chiral stationary phase (Chiralcel® OJ™ column (Daicel Corporation, Tokyo, Japan), diameter 21 mm, length 250 mm; mobile phase: n-hexane/ethanol 80:20 + 0.5% diethylamine, flow: 20 mL/min). The desired active R-enantiomer of SCH900776 eluted faster (retention time: 10:04 min) than the inactive S-enantiomer (retention time: 13:07 min). LY2603618 was purchased from Selleckchem (Houston, TX, USA; cat. no. S2626).
Cell culture
HCT116 p53+/+ and p53−/− cells were a kind gift from B. Vogelstein. 30 The cells were cultivated in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich, D6429, Darmstadt, Germany) medium supplemented with 5% fetal bovine serum (FBS; Sigma-Aldrich, F0804) at 37°C. MDA-MB-231 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and were cultivated in DMEM medium supplemented with 10% FBS at 37°C with 5% CO2.
Proliferation assays
HCT116 cells were transfected with the following small interfering RNAs (siRNAs; Santa Cruz Biotechnology, Dallas, TX, USA): CTRL A (sc-37007), BRCA1 (sc-29219), CDK12 (sc-44531), CDK13 (sc-72836), and CDK12_2 (Sigma-Aldrich, SIHK0490) using Lipofectamine RNAiMAX (Invitrogen, 13778150, Carlsbad, CA, USA). Viable cells were counted after 24 h and equal cell concentrations were seeded into 96-well plates. After an additional 24 h, cells were treated with a CHK1 inhibitor, either SCH900776 or LY2603618, in dimethyl sulfoxide (DMSO) for 6 days. The medium was exchanged after 48 and 96 h, and fresh inhibitors were added to the medium at these time points. For each siRNA, the cell viability was assessed with the CyQuant NF Cell Proliferation Assay Kit (Invitrogen) and normalized to the relative growth of cells treated with DMSO. All experiments were performed three times in triplicates.
MDA-MB-231 cells were seeded at equal concentrations into six-well plates. After 24 h, they were exposed to DMSO, along with either 0.3 or 1 µM SCH900776. After 72 h, the cells were trypsinized and counted with a hemocytometer. Cell viability was normalized to relative growth of cells treated with DMSO for each short hairpin RNA (shRNA). The experiments were performed three times in duplicates.
Western blot
Cells were lysed in lysis buffer (100 mM Tris, pH 7.4, 1% sodium dodecyl sulfate (SDS), 10% glycerol), sonicated, and protein concentrations were assessed by the bicinchoninic acid (BCA) assay. Laemmli buffer (3×) was then added, and lysates were boiled for 5 min at 100°C. Cell lysates were separated with electrophoresis employing 8%–15% gels and then wet blotted to a nitrocellulose membrane (GE Healthcare, Amersham, #10600008, Little Chalfont, UK). Individual proteins were detected with specific antibodies: BRCA1 (Santa Cruz, sc-6954), CDK12 (Cell Signaling, #11973, Danvers, MA, USA), CDK13 (rabbit serum produced in-house), Cyclin K (Santa Cruz, sc-376371), p53 Pantropic Ab-6 (Millipore, OP43, Billerica, MA, USA), Cyclin T1 (Santa Cruz, sc-8127), CHK1 (Cell Signaling, #2360), CHK1-pSer296 (Cell Signaling, #2349), PARP (Cell Signaling, #9542), γH2AX pSer 139 (Biolegend, 613402, San Diego, CA, USA), p21 (Santa Cruz, sc-397), p27 (Santa Cruz, sc-528), pRb (Cell Signaling, #9309), and pRb-pSer780 (Cell Signaling, #8180). Anti-rabbit and anti-mouse secondary horseradish peroxidase (HRP)-linked antibodies were obtained from GE Healthcare (NA934V, NA931V), and anti-goat antibody was obtained from Sigma-Aldrich (A5420). The immunoreactive bands were visualized using the Western blot Luminol reagent (Santa Cruz Biotechnology, SC-2048).
Reverse transcription–polymerase chain reaction
HCT116 cells were transfected with control or specific siRNAs using lipofectamine RNAiMAX (Invitrogen, 13778150). After 72 h, cells were harvested, and total RNA was isolated with RNAzol (Molecular Research Centre, RN190, Cincinnati, OH, USA). Reverse transcription (RT) and quantitative polymerase chain reaction (qPCR) were performed according to the method described by Blazek et al. 16 Changes in gene expression were calculated using the comparative threshold cycle method with Hypoxanthine Phosphoribosyltransferase 1 (HPRT) to normalize for variations in RNA input. The following primers were used for PCR: CDKN1A—forward: CTGGAGACTCTCAGGGTCGAAA and reverse: GATTAGGGCTTCCTCTTG and HPRT—forward: 5′-CCAGACAAGTTTGTTGTAGGATATGCCCTTGAC-3′ and reverse: 5′-ACTCCAGATGTTTCCAAACTCAACTTGAACTCTC-3′.
Plasmids
The pLKO.1 shBRCA1-2 and pLKO.1 shBRCA1-4 plasmids, which were used to generate stable BRCA1 knockdown cell lines, were part of the MISSION library (Sigma-Aldrich; construct numbers TRCN0000244985 and TRCN0000244987). The lentiviral packaging plasmids pMD2.G and psPAX2 were purchased from Addgene (Cambridge, MA, USA; Plasmids numbers 12259 and 12260).
Generation of MDA-MB-231 shBRCA1 cell lines
Lentiviral transduction was used to generate MDA-MB-231 cell lines that harbor a stable shRNA knockdown of BRCA1. Lentivirus production and transduction was performed according to the method described by Tiscornia et al. 31 Briefly, lentiviruses were generated by co-transfecting 293T cells with 4 µg of pMD2.G, 7 µg of psPAX2, and 9 µg of a lentiviral plasmid of interest using the CaPO4 precipitation method. Next, 6–8 h post-transfection, cells were washed with pre-warmed phosphate-buffered saline (PBS) and the medium was changed. Supernatant containing lentiviruses was collected 48 h later and supplemented with 4 µg/mL polybrene (Sigma-Aldrich, 107689). Target cells were transduced at multiplicities of infection (MOIs) of 1–10. The medium was changed 24 h post-transduction, and the cells were selected with 1 µg/mL puromycin. Resistant colonies were evaluated for expression of shRNA and the consequent reduction in BRCA1 protein levels.
Clonogenic assay
MDA-MB-231 cells were seeded at equal concentrations into six-well plates (150 cells/well) and, after 24 h, treated with 0, 0.3, or 1 µM SCH900776 for 14 days. Medium was exchanged and fresh inhibitors were added every 3 days. After 2 weeks of treatment, the cells were fixed with 4% paraformaldehyde and stained with Gram I Solution (PENTA s.r.o., cat. no. 14600-11000, Prague, Czech Republic).
Cell cycle analysis
HCT116 or MDA-MB-231 cells were trypsinized, washed in PBS, and fixed with ice-cold 70% ethanol overnight. Cells were then washed in PBS and stained with Vindelov solution (1M TrisHCl, pH 8.0; 0.1% Triton-X100; 10 mM NaCl; propidium iodide, 50 μg/mL; RNAse A 50 Kunitz U/mL) for 30 min at 37°C.
Xenograft model
All animal procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee. Tumor xenografts were generated by injecting 2 × 106 MDA-MB-231 parental or shBRCA #4 breast cancer cells, in PBS with a final volume of 100 μL, into the left mammary fat pads of Ctrl: SHO-PrkdcSCID HrHr mice (Charles River Laboratories, Wilmington, MA, USA). In total, 12 mice were injected with MDA-MB-231 parental or shBRCA #4 cells and half of them (six mice) from particular transplantation was treated with the vehicle or CHK1 inhibitor SCH900776. Drug treatment began once a tumor had reached 0.03 cm3. The CHK1 inhibitor SCH900776 was dissolved in 20% Kolliphor ELP (Sigma-Aldrich, 30906) and administered intraperitoneally at a final concentration of 25 mg/kg for 5 days. This dose was selected based on a previously published study that investigated the same CHK1 inhibitor. 6 Two-dimensional calipers were used to measure tumor volumes during and after the treatment period, and volume was calculated based on the equation: π/6 × length × width2. 9 Data were normalized to the starting tumor volume of 0.03 cm3. The whole experiment was carried out in two independent replicates. Increase in tumor volume after the treatment was statistically analyzed by Student’s t-test.
Statistical analysis
All data were statistically analyzed and visualized using Prism5 (GraphPad Software, Inc., La Jolla, USA). Results were expressed as mean with standard error of the mean (SEM). Viability assays data were fitted to sigmoidal dose response curve and were analyzed with analysis of variance (ANOVA) test, and data from other experiments were analyzed by Student’s t-test. Symbols used to express statistical significance are as follows: *p ≤ 0.05; **p ≤ 0.01; and ***p ≤ 0.001.
Results
CDK12 and BRCA1 downregulation sensitizes HCT116 cells to CHK1 inhibition irrespective of p53 status
CDK12 regulates BRCA1 expression, and a loss of BRCA1 results in increased DNA damage and replication stress.16,27 Thus, we hypothesized that cells with depleted BRCA1 or CDK12 should be extensively dependent on the S-phase-related function of CHK1. We tested how the CHK1 inhibitors SCH900776 and LY2603618 affect the proliferation of BRCA1- and CDK12-silenced tumor cells. Whether the functional status of p53 influences the antiproliferative effect of CHK1 inhibitors remains an open question, so we employed a pair of HCT116 cell lines with p53 null and p53 WT status. 32 In addition to CDK12 we validated the effects of CHK1 inhibitors on cells with silenced CDK13, another kinase binding Cyclin K that can phosphorylate RNAPII, 33 but that has no impact on BRCA1 levels.
We performed 6-day proliferation assays to assess the relationship between CDK12 or BRCA1 deficiency and CHK1 inhibition (Figure 1). The siRNA-transfected cells were treated with different concentrations of CHK1 inhibitor SCH900776. We used two distinct siRNAs against CDK12, a pool of three siRNAs (CDK12), and a single sequence different from the other three (CDK12_2). CDK12 and BRCA1 silencing significantly sensitized both p53+/+ and p53−/− cell lines to the CHK1 inhibitor SCH900776. In contrast, CDK13 silencing had no effect on sensitivity to SCH900776 (Figure 1(a) and (b)). To avoid the risk that the effect of SCH900776 on cell proliferation is compound-specific (i.e. that it employs unknown off-target effects), we tested the effect of another CHK1 inhibitor, LY2603618, which has a different chemical structure, 34 using the same setup as in Figure 1(a) and (b). This experiment provided similar results as those obtained when SCH900776 was used (Figure 1(c) and (d)). It is important to note that sensitivity to both tested CHK1 inhibitors was independent of the p53 status. In order to demonstrate that effect of CHK1 inhibitor is not limited only to the colorectal cell line HCT116, the same type of experiment as in Figure 1(a) was performed with an ovarian cancer cell line OVSAHO, which bears p53 mutation and wild-type BRCA1. 35 Sensitization to CHK1 inhibitor after BRCA1 and CDK12 silencing was observed similar to HCT116 cell line (Supplementary Figure S1).

Downregulation of CDK12 or BRCA1 sensitizes HCT116 cells to CHK1 inhibitors. Six-day survival curves of (a, c) HCT116 p53+/+ or (b, d) HCT116 p53−/− cells transfected with various siRNAs (CTRL, CDK12, CDK13, and BRCA1) and treated with either the CHK1 inhibitor (a, b) SCH900776 or (c, d) LY2603618. Cell viability for each siRNA-treated cell line was assessed by the CyQuant NF kit and normalized to the relative growth of cells treated with DMSO. Error bars represent SEM for three independent experiments. CDK12- and BRCA1-silenced cells are sensitive to CHK1 inhibitors (p < 0.001, ANOVA). (e) The effective knockdown of indicated proteins after siRNA transfections was examined by Western blot analysis. HCT116 p53+/+ and HCT116 p53−/− cells were transfected with various siRNAs and protein levels were assessed by Western blot analysis after 72 h. The protein level of Cyclin T1 was used as a loading control.
The effective downregulation of CDK12, CDK13, cyclin K (associating partner of CDK12 and CDK13), and BRCA1 in the cells was verified by Western blot analysis (Figure 1(e)). As expected, the protein levels of cyclin K and BRCA1 decreased after CDK12 downregulation, which corresponds to our previous observations.16,20. Cyclin T1, an associating partner of CDK9 kinase involved in transcription elongation, was used as a loading control.
BRCA1 or CDK12 depletion coupled with CHK1 inhibition induces p21-dependent proliferation block
We also investigated endogenous DNA damage, apoptosis, autophagy, and cell cycle status to gain more insight into the molecular mechanisms responsible for the enhanced cytostatic effect of CDK12 or BRCA1 downregulation coupled with CHK1 inhibition. HCT116 cells were depleted of CDK12, CDK13, or BRCA1 by siRNAs and were then, 24 h post-transfection, exposed to a CHK1 inhibitor (SCH900776) for an additional 96 h. Samples were collected and assessed for the DNA damage marker yH2AX 36 and CHK1 autophosphorylation at serine 296, an indication of activated CHK1 kinase. 14
BRCA1 and Cyclin K levels once again decreased following CDK12 knockdown (Figure 2(a)). As expected, treatment with CHK1 inhibitor SCH900776 led to a noticeable decrease in the detected p-S296 CHK1 signal at all tested conditions (Figure 2(a)), and also induced CHK1 degradation in a dose-dependent manner, which is consistent with published research. 14 Importantly, the yH2AX signal, which reflects the amount of endogenous DNA damage, significantly increased after SCH900776 treatment. This effect was exacerbated in CDK12- and BRCA1-depleted cells, but not in CDK13 cells (Figure 2(a)).

Impact of CDK12 and BRCA1 downregulation on DDR, apoptosis and cell cycle. (a) The effective knockdown of various proteins after siRNA transfection, CHK1 inhibition and DNA damage induction was assessed by Western blot analysis. HCT116 p53+/+ cells were transfected with control or specific siRNAs (CTRL, CDK12, CDK13, and BRCA1) and 2 days post-transfection cells were treated with 0, 0.3, or 1 μM CHK1 inhibitor SCH900776 for an additional 96 h. The protein levels of the studied proteins were elucidated by Western blot with indicated antibodies. The protein level of Cyclin T1 was used as a loading control. (b) Status of cellular factors participating in regulation of apoptosis and cell cycle. The protein levels of PARP, a marker of late apoptosis, tumor suppressor p53, and the cell-cycle regulating proteins p21, p27, pRb, and pRB-pSer780 were elucidated by Western blot with indicated antibodies. The protein level of Cyclin T1 was used as a loading control.
Together, these results verified that our experimental setting was successfully able to inhibit CHK1 and confirmed our hypothesis that CHK1 is necessary for the effective repair of endogenous DNA damage, especially in cells lacking functional components of DDR such as CDK12 or BRCA1.
Next we focused on induction of apoptosis, which was examined by the presence of cleaved PARP and Caspase 3, a commonly used markers of apoptosis. Surprisingly, SCH900776 treatment of CDK12-depleted cells only moderately increased PARP-1 cleavage (Figure 2(b)). In addition, no cleaved Caspase 3 was detected after transfection with any siRNAs or after administration of CHK1 inhibitor (Supplementary Figure S2). Furthermore, we were interested whether other types of cell death that might play a role in viability of CDK12-silenced cells, therefore the protein levels of specific autophagy markers were evaluated. As depicted in Supplementary Figure S2, no significant change in the protein level of Beclin, factor associated with vesicle-trafficking during autophagy, after CHK1 inhibitor administration in combination with any of the tested siRNAs was identified. Also, no detectable change in protein level of the cleaved product of LC3B protein was detected in any tested conditions.
Because decreased cell viability cannot be entirely explained by apoptosis or autophagy, we elucidated cell cycle progression. To corroborate this further, we checked the status of the p21 (Cip1/Waf1), a protein that is a prominent inhibitor of CDKs, can induce cell cycle blockade, and is known to respond to CHK1 inhibition. 14 Indeed, p21 protein levels increased proportionally to CHK1 inhibition under all tested conditions, with BRCA1-depleted cells showing the strongest induction (HCT116 p53+/+) (Figure 2(b), lanes 3 and 12). Interestingly, a robust induction of p21 was also observed upon CDK12 downregulation, even without CHK1 inhibitor treatment (Figure 2(b), lanes 4–6). The p21 induction was detected in HCT116 p53−/− cells as well (Supplementary Figure S3). On the contrary, the expression levels of a related cellular inhibitor of CDKs, the p27 protein, did not change following CHK1 inhibitor treatment, implying a specific induction of p21 in response to CDK12 downregulation (Figure 2(b)).
In addition to regulating G1/S progression through CDK inhibition, p21 also induces the degradation of another cell cycle regulator, the Retinoblastoma protein (pRb). 37 Therefore, we evaluated the levels and phosphorylation status of pRb. Silencing of CDK12 led to degradation of pRb regardless of CHK1 inhibition, and a moderate effect was also observed in BRCA1 depleted cells after treatment with 1 µM of SCH900776 (Figure 2(b)). In addition, the cell cycle status was examined by propidium iodide staining followed by flow cytometry. Depletion of CDK12 together with administration of CHK1 inhibitor led to significant prolonged G1 phase and shortened S phase of the cell cycle in comparison to CTRL-, CDK13-, or BRCA1-silenced cells (Supplementary Figure S4).
Based on these data, we conclude that the enhanced cytostatic effect of CHK1 inhibition in CDK12- or BRCA1-depleted HCT116 p53+/+ cells is a result of increased DNA damage, which leads to a robust induction of p21 and delayed cell cycle progression.
BRCA1 depletion sensitizes MDA-MB-231 cells to CHK1 inhibition
Next we tested whether BRCA1 deficiency can sensitize other cancer models to the chemical inhibition of CHK1 kinase. Since BRCA1 deregulation or loss-of-function mutations are common characteristics of ovarian and breast cancers,38,39 we chose to manipulate MDA-MB-231 breast cancer cells, which have mutated form of p53 and normal BRCA1 status, prior to the administration of a CHK1 inhibitor. The therapeutic potential of CHK1 inhibitors has been already tested in triple-negative breast cancer (TNBC) cell lines and pre-clinical mouse models, but the status of BRCA1 has not been modified. 11
First, we generated two MDA-MB-231 cell lines with stable expression of BRCA1 shRNA (MDA-MB-231 shBRCA1 #2 and #4). Successful BRCA1 depletion at the protein and mRNA level was confirmed by Western blot and RT-PCR, respectively (Figure 3(a)). The effect of BRCA1 downregulation and CHK1 inhibition on cellular viability was assessed by a survival assay that employed the same setup as for HCT116 cells (Figure 3(b)). Treatment with 1 µM of SCH900776 led to a severe reduction in cell viability, decreasing the cell counts of both depleted cell lines by 70% when compared to the parental non-transfected cells (Figure 3(b)). We examined the effect of CHK1 inhibition on cell viability in MDA-MB-231 (shBRCA1 #2 and #4) cell lines further by performing a clonogenic survival assay, which better reflects the effects of long-term exposure (Figure 3(c)). After 14 days of cultivation, we noticed that the colonies of both untreated shBRCA1 cell lines were larger than those of the parental cell line, suggesting faster cell cycle progression and higher mitotic potential. This is in line with the observation that BRCA1 loss accelerates the growth of cancer cells. 40 However, treatment with a CHK1 inhibitor (1 μM) noticeably reduced colony size in both shBRCA1 cell lines and also led to dramatic decrease in cell counts (Figure 3(c))
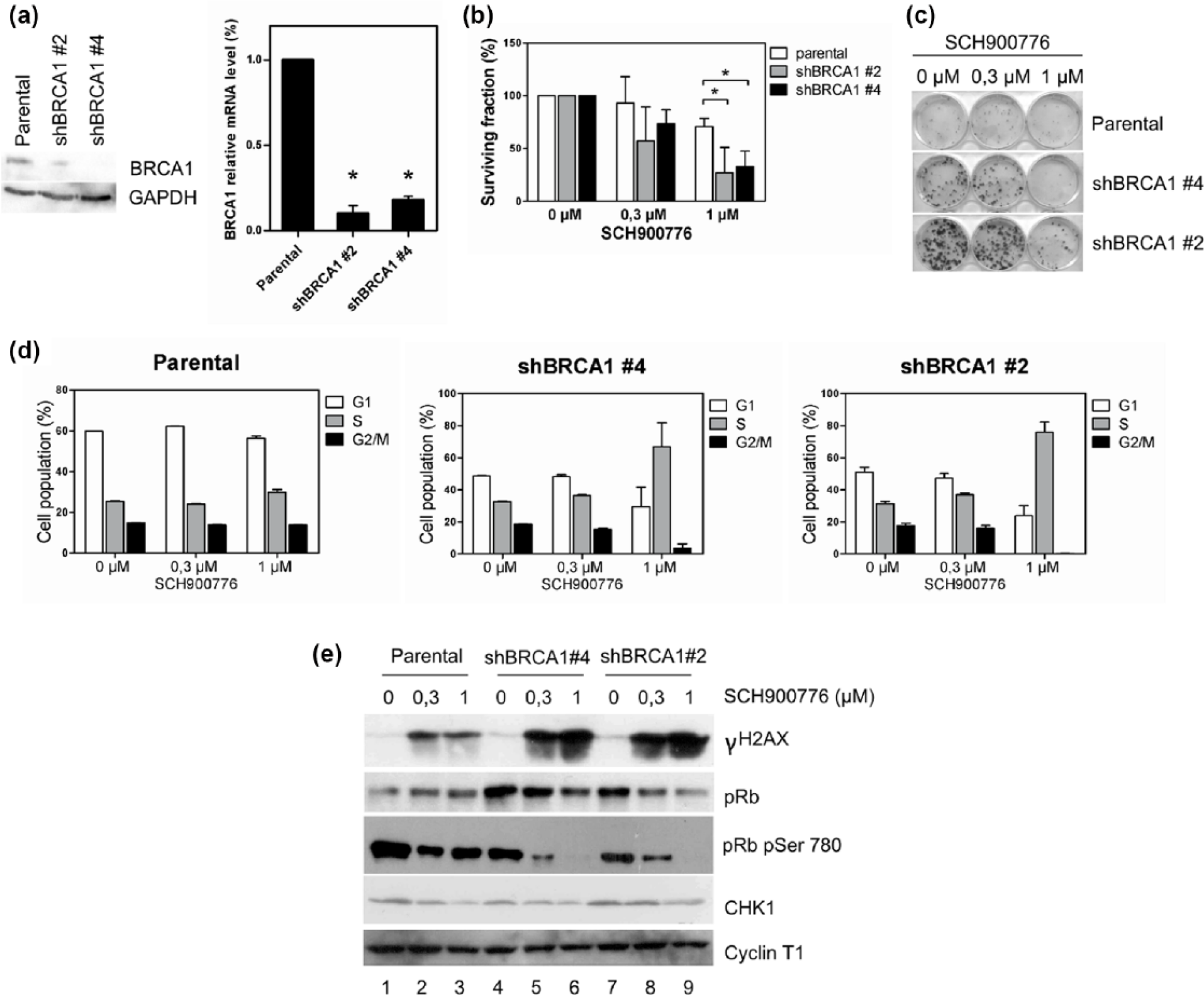
Downregulation of BRCA1 sensitizes MDA-MB-231 cells to CHK1 inhibitor. (a) BRCA1 protein levels were evaluated in shBRCA1 #2 and shBRCA1 #4 MDA-MB-231 cell lines by Western blot analysis. GAPDH was used as a loading control. The mRNA levels of BRCA1 in these cell lines were measured by RT-qPCR (p < 0.05, Student’s t-test). These experiments confirmed effective BRCA1 downregulation by shRNAs in these cell lines. (b) CHK1 inhibition reduced the viability of MDA-MB-231 cells expressing shRNAs against BRCA1. The graphs show the results of survival assays of parental, shBRCA1 #2 and shBRCA1 #4 MDA-MB-231 cells treated with 0, 0, 3, or 1 μM CHK1 inhibitor SCH900776 for 3 days. For each cell line, the cell numbers were normalized to the relative growth of cells treated with DMSO. Error bars represent SEM for three independent experiments (p < 0.05, Student’s t-test). (c) A 14-day clonogenic assay showed that the combination of BRCA1 silencing and CHK1 inhibition reduces cell viability. MDA-MB-231 cell lines were seeded and treated with the indicated SCH900776 concentrations. The experiment was performed three times in duplicates. BRCA1 silencing combined with CHK1 inhibition reduces cell viability. (d) CHK1 inhibition strongly affected the cell cycle progression of BRCA1-silenced MDA-MB-231 cell lines. Cells were cultivated and treated as described in (b). Cell cycle progression was evaluated by the incorporation of propidium iodide followed by flow cytometry. Error bars represent SEM from three experiments. (e) The combination of BRCA1 silencing and CHK1 inhibition induces a stronger activation of DDR. Cell were prepared and treated as described in (b). The activation of DDR and inhibition of CHK1 by SCH900776 were validated by Western blot with antibodies against γH2AX pSer139 and CHK1, respectively. The dose-dependent degradation of total pRb and pRb phosphorylated on serine 780 following CHK1 inhibition was examined by Western blot. Cyclin T1 was used as a loading control.
Moreover, the 3-day inhibition of CHK1 had no apparent effect on cell cycle in the parental MDA-MB-231 cell line, whereas the shBRCA1 clones experienced a prominent increase in the S-phase cells, suggesting major proliferation arrest (Figure 3(d)).
We then tested how BRCA1 downregulation enhances endogenous DNA instability. A rather moderate, yet reproducible, increase in γH2AX levels was observed in both shBRCA1 cell lines in comparison to the parental cells (Figure 3(e), lanes 1, 4, and 7). Interestingly, CHK1 inhibition led to a dramatic increase in the yH2AX signal in both shBRCA1 cell lines, while the parental cells only demonstrated a moderate dose-dependent γH2AX response (Figure 3(e)). As was earlier observed in the HCT116 cells, CHK1 inhibition led to a reduction in CHK1 levels and a dose-dependent decrease in total and phosphorylated pRb levels (Figure 3(e)).
In summary, BRCA1 downregulation also sensitizes MDA-MB-231 breast cancer cells to CHK1 inhibitors. As in HCT116 cells, treatment with a CHK1 inhibitor induced excessive DNA damage followed by arrest in the S-phase.
Xenograft mouse model
Based on our in vitro results, we decided to assess the in vivo therapeutic effects of CHK1 inhibitors by employing a mouse orthotopic xenograft model of the MDA-MB-231 parental and shBRCA1 #4 human breast carcinoma cells in the fat pads of SHO-PrkdcSCID HrHr mice. Mice were continuously monitored for the development of a primary xenograft tumor and sacrificed when tumors reached 10% of body weight. Besides the observation that animals transplanted with either parental or shBRCA1 #4 MDA-MB-231 cells all formed tumors (100% tumor growth), mice injected with shRBCA1 cells showed a slightly higher proportion of tumor volume, reflecting faster growth of shBRCA1 cells (Figure 3(c)). Furthermore, the growth rate and final size of the tumors differed distinctly between populations after CHK1 inhibitor treatment. The parental MDA-MB-231 control cells formed significant tumors over the course of 44 days, but treatment with a CHK1 inhibitor did not significantly affect the size of the tumors when compared to control animals (Figure 4(a)). In contrast, the animals injected with shBRCA1 MDA-MB-231 cells developed significantly larger tumors than the control group, and treatment with a CHK1 inhibitor significantly decreased tumor size (Figure 4(b)).

Inhibition of CHK1 prevents tumor growth in vivo. The CHK1 inhibitor SCH900776 decreased tumor growth. The (a) parental and (b) shBRCA1 MDA-MB-231 cells were transplanted into the mammary pads of SCID mice. When tumor mass reached a volume of 0.03 cm3, mice were treated with either a vehicle solution of 20% Kolliphor ELP or SCH900776 25 mg/kg/day dissolved in the same 20% Kolliphor solution, every day for 5 days. The growth of tumor mass was then monitored over set time periods. Each data point represents the mean increase in tumor volume after the beginning of treatment and error bars represent SEM, where n for each cohort was six animals (p < 0.001, Student’s t-test for shBRCA1 SCH900776 vs shBRCA1 vehicle).
Discussion
CHK1 inhibitors represent a promising cancer therapy approach. 3 Since the anti-cancer effect of CHK1 inhibitors is potentiated by DNA damaging drugs, we hypothesized that impaired DDR will have a similar synergistic effect. In our previous study, we demonstrated that CDK12 regulates the transcription of certain DDR genes, particularly HR genes (including BRCA1), and is necessary for maintaining genomic stability. 16 In line with this observation, the loss of either BRCA1 or CDK12 is a prerequisite for sensitizing cancer cells to PARP1/2 inhibitors.21,41 In this study, we demonstrate that the loss of BRCA1 or CDK12 also potentiates the anti-proliferative effect of CHK1 inhibitors.
Our results show that the anti-proliferative effect of CHK1 inhibitor treatment combined with BRCA1 or CDK12 deficiency is comparable in both cell lines regardless of p53 status. Previous reports have shown that CHK1 inhibition leads to increased DNA damage by measuring the induction of γH2AX pSer139 in HCT116 cells.6,14 The strongest effect was obtained when CHK1 inhibition was combined with CDK12 silencing. Interestingly, in contrast to the results obtained in HeLa cells, 16 we did not observe increased γH2AX Ser139 phosphorylation upon CDK12 depletion in HCT116 cells. This may be partially due to differences between these cell lines. However, we observed robust γH2AX pSer139 induction in the MDA-MB-231 BRCA-silenced cells after CHK1 inhibition. CHK1 inhibition was previously reported to induce the cell cycle regulator p21. 14 We observed a particularly robust p21 increase in BRCA1-silenced cells. CDK12 silencing (regardless of CHK1 inhibition) resulted in increased apoptosis, which was consistent with result obtained from CDK12 inhibition. 42 Based on the data obtained from MDA-MB-231 cells, one could speculate that CHK1 inhibitors trigger increased DNA damage and replication stress during the S-phase of the cell cycle, which is incompatible with cell proliferation or survival. Moreover, the potential of CHK1 inhibitors to weaken tumor growth has been reported by several groups, but, as of yet, the synergy between impaired CDK12 function and CHK1 inhibition to counteract tumor progression has not been investigated.6,9,11
It has been demonstrated that CDK12 regulates the expression of DDR genes including CHK1. 20 Importantly, high-grade serous ovarian tumors bearing CDK12 mutations have reduced CHK1 expression. 20 Therefore, we speculate that viability of CDK12-depleted cells rely extensively on the residual activity of CHK1 making these cells sensitive to lower doses of CHK1 inhibitors.
Several studies have demonstrated that CDK12 regulates the expression of DDR genes (BRCA1, FANCI, FANCD2, and ATM).16,20,42 Nevertheless, the precise mechanism of this regulation has not yet been described. CDK12 loss might downregulate DDR genes directly through the role of CDK12 in the transcription of these genes, as has been suggested by numerous studies.16,19,20,42,43
In addition, a recent publication reported that genomic instability in ovarian tumors with a loss of CDK12 has a specific pattern of defective HR caused by BRCA1/2 deficiency suggesting that CDK12 functions in additional parts of the DNA repair machinery. 44 From the clinical point of view, about 9% and 13% of unselected women with newly diagnosed triple-negative breast cancer and ovarian cancer (respectively) have an inheritable BRCA1 mutation. If metastatic, these patients have generally very unfavorable prognosis and currently are candidates for targeted drug therapy, such as PARP inhibitors.24–26 We confirmed the additive effect of BRCA1 loss-of-function and CHK1 on cell proliferation in vitro and in vivo. A xenograft model was employed to evaluate whether CHK1 inhibition confers an anti-proliferative effect on tumor growth in vivo. CHK1 inhibitor administration had no substantial impact on the parental MDA-MB-231 cells, which was in sharp contrast to significant decrease in tumor mass observed in BRCA1-silenced cells receiving CHK1 inhibitors (Figure 4). Recent studies have clearly shown that patients can develop a resistance to PARP1/2 inhibitors over the course of PARP inhibitor therapy. 45 Since many factors, such as HR and NHEJ status, as well as the level, activity, or intracellular concentration of PARP proteins, can influence the efficacy of PARP inhibitors, it is vital to identify different conditions that can either re-sensitize tumor cells to PARP inhibitors or enable the use of additional strategies that target the systems necessary for cell survival to treat resistant tumors. The anti-proliferative effect of CHK1 inhibitors in BRCA1-deficient (HR compromised) tumor cells differs from that of PARP1 inhibitors; hence, it seems rational to investigate how using CHK1 inhibitors as a second round of therapy will affect patients who have tumors resistant to PARP inhibitors due to the restoration of HR.
In summary, we have found that CHK1 inhibition is a promising strategy for targeting BRCA1- or CDK12-deficient cells. We propose that BRCA1 and CDK12 deficiency should be considered a CHK1 sensitivity biomarker candidate. The cell cycle arrest triggered by recently developed specific CDK12 inhibitors is in line with our presented observations. 42 Moreover, combination therapy with PARP1/2 inhibitors and a CDK12 inhibitor conferred a strong anti-proliferative effect in breast cancer cells. 41 Our results provide promising evidence for the combinatory effect of CDK12 and CHK1 inhibitors in treating cancer patients.41–43
Footnotes
Acknowledgements
The authors certify that they have NO affiliations with, or involvement in, any organization or entity with any financial or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript. The authors would like to thank Dr Sabina Sevcikova and members of the J. Kohoutek laboratory for critical discussion and their suggestions during the preparation of this manuscript, Dr Jiri Jarkovsky for statistical analyses, Prof. Jiri Bartek for helpful discussion, D. Blazek for providing us with OVSAHO cells and Dr Miroslav Machala for the sharing antibodies that were used in the Western blot analyses.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Internal Grant Agency of the Ministry of Health of the Czech Republic, grant NT14599-3/2013 (M.S. and J.K.); the Ministry of Health of the Czech Republic, grant 16-34152A (J.K.); the Ministry of Agriculture, grant RO0517; the European Regional Development Fund, project no. LQ1605 from the National Program of Sustainability II (MEYS CR) (K.S., K.P., and O.H.); the Ministry of Health of the Czech Republic, grant 15-33999A (M.S., K.P., and K.S.); CZ-OPENSCREEN: National Infrastructure for Chemical Biology, LM2015063 (K.P.); the Czech National Program of Sustainability, LO1304 (J.K. and M.M.); and the Norwegian Financial Mechanism 2009-2014 Project, Contract 7F14061 (J.K.).
Supplementary material
Supplementary material is available for this article online
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.