
Abstract
Cancer is among the leading causes of death worldwide, and the number of new cases continues to rise. Despite recent advances in diagnosis and therapeutic strategies, millions of cancer-related deaths occur, indicating the need for better therapies and diagnostic strategies. Mitochondria and metabolic alterations have been recognized as important for cancer progression. However, a more precise understanding of how to manipulate mitochondria-related processes for cancer therapy remains to be established. Mitochondria are highly dynamic organelles which continually fuse and divide in response to diverse stimuli. Participation in the aforementioned processes requires a precise regulation at many levels that allows the cell to couple mitochondrial activity to nutrient availability, biosynthetic demands, proliferation rates, and external stimuli. The many functions of these organelles are intimately linked to their morphology. Recent evidence suggests an important link between mitochondrial morphology and disease, including neurodegenerative, inflammatory diseases and cancer. Here, we review recent advances in the understanding of mitochondrial dynamics with a special focus on its relationship to tumor progression.
Introduction
Mitochondria are often considered the cell’s powerhouse, not only due to their essential role in the production of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) but also because of their crucial role in other biochemical pathways such as pyrimidine and purine biosynthesis, heme synthesis, the regulation of nitrogen balance in the urea cycle, gluconeogenesis, ketone body production, and lipid degradation and elongation.1,2 Emerging evidence has also demonstrated that mitochondria play an important role in the regulation of cell signaling, either serving as a scaffold for protein–protein interactions or by regulating the levels of intracellular messengers such as Ca2+ or reactive oxygen species (ROS).
Our understanding of mitochondrial dynamics, biogenesis, and degradation has dramatically increased in recent years and evidence has shown that alterations in these processes are linked to diverse pathological states including cancer. Mitochondrial integrity is fundamental for survival in response to energetic, environmental, and genotoxic stressors. Despite a clear understanding of the importance of mitochondrial dynamics, its precise role in the clinical outcome of cancer patients remains to be established. In this regard, increasing evidence suggests a potential utility of diverse mitochondria-related parameters for cancer diagnosis and treatment, but the role of mitochondria in cancer development or progression is not simple. Here, we describe the current knowledge of the processes regulating mitochondrial shape and function and review recent studies on their role in tumorigenesis and cancer progression, focusing on the potential use of mitochondrial biomarkers for cancer detection and prognosis as well as on the possibility of targeting mitochondrial dynamics for cancer therapy.
Mitochondrial fusion and fission
Our view of mitochondrial dynamics has dramatically changed in the past years. Once considered to be a rigid structure, mitochondria are now known to be organelles that migrate throughout the cell, fuse, divide, and undergo regulated turnover through mitochondrial autophagy or mitophagy. Mitochondria continually divide and fuse, even in resting cells where the mitochondrial network is formed by a mixture of long and interconnected tubules.3–5 In some cells, they fuse together in a connected network, whereas in other cells or under different circumstances, mitochondria convert into a large number of small fragments. Fibroblast mitochondria, for example, are usually long filaments, whereas hepatocyte mitochondria are more uniformly spheres or ovoids. 3 The many functions of mitochondria are intimately linked to their morphology, and there is a delicate balance between the fusion and fission events, which allows proper mitochondrial function in the cell.
Unopposed fission causes extensive mitochondrial fragmentation, which has been generally associated with cellular metabolic dysfunction and disease. However, continuous fusion results in a hyperfused network, which counteracts metabolic insults, preserves cellular integrity, and protects against autophagy. 6 Thus, balanced fusion and fission events shape mitochondria to meet cellular metabolic demands and to regulate the removal of damaged organelles. Besides being involved in the maintenance of normal cell functions as the distribution of mitochondria during cell growth, cell division, and differentiation, mitochondrial dynamics has recently been considered as a cornerstone for the maintenance of cellular fitness since alterations in fusion and fission events have been closely linked to states of health and disease (Figure 1).5,6

Mitochondrial morphology and cellular processes that have been related to them. Mitochondrial dynamics are the result of a delicate balance between mitochondrial fusion (characterized by the presence of long interconnected tubules) and fission (characterized by small, fragmented mitochondria). Diverse cellular functions have been associated with mitochondrial morphology, as shown in the figure and reviewed in the text.
The molecular machinery regulating mitochondrial dynamics comprises large dynamin-like guanosine triphosphatases (GTPases) mediating the fusion and fission of both outer mitochondrial membrane (OMM) and inner membrane (IMM; Figure 2). OMM fusion is regulated by mitofusins 1 and 2 (MFN1 and MFN2), which are required for the maintenance of a reticular mitochondrial network in cells. 7 Mitofusins function through homo- and heterotypic interactions, and their synthesis is regulated by transcriptional and post-transcriptional mechanisms (Figure 2). 6 The transcriptional mitochondrial coactivators phosphatidylglycerol phospholipase C-1-alpha and beta (PGC-1-α and β), which regulate mitochondrial biogenesis and the expression of the respiratory complex machinery, have also been involved in the regulation of mitochondrial dynamics through control of gene expression.8,9 In this regard, treatment of mice with resveratrol, a known activator of the protein deacetylase sirtuin 1 (SIRT1), decreased PGC-1α acetylation and increased its activity. These effects led to the induction of mitochondrial biogenesis and increased mitochondrial size in skeletal muscle, conferring metabolic advantages like increased aerobic capacity in vivo. 6 Also, PGC-1-β has been proposed to induce mitochondrial fusion by increasing the transcription of Mfn2 through estrogen-related receptor alpha (ERRα) coactivation. 9
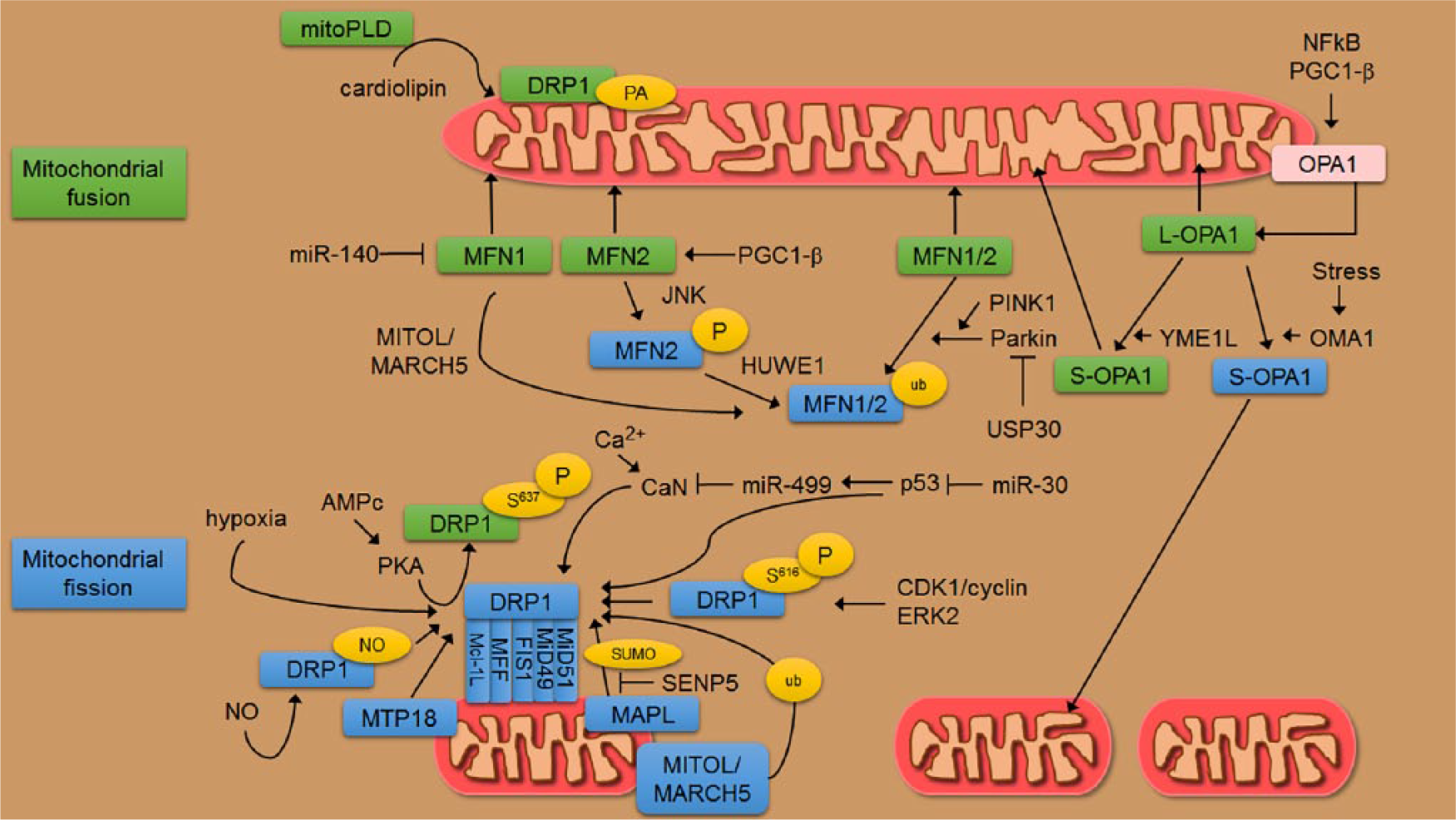
Molecular machinery involved in the shaping of mitochondria. Mitochondrial shape is regulated by a family of GTPases with structural homologies to dynamin proteins. Mitofusins are the GTPases required for OMM fusion and they are regulated by ubiquitylation and phosphorylation. The E3-ubiquitin ligase PARKIN marks both MFN1 and MFN2 with ubiquitin (ub) regulating their activity and levels. However, the deubiquitylase USP30 antagonizes PARKIN activity promoting mitochondrial fusion. PINK1 is a kinase able to phosphorylate ubiquitin chains on OMM proteins including MFN2 which can then recruit PARKIN. Phosphorylation of MFN2 by JNK in response to cellular stress signals the recruitment of the E3-ubiquitin ligase HUWE1, triggering MFN2 degradation by the proteasome. Also, miR-140, which can be induced by oxidative or genotoxic insults, negatively regulates MFN1 translation. The transcriptional coactivator PGC-1-β has been proposed to regulate mitochondrial fusion via Mfn2 and other mitochondrial genes like OPA1. MitoPLD is a member of the phospholipase D family which is bound to the outer membrane and converts cardiolipin to phosphatidic acid. This catalytic activity is required for mitochondrial fusion. In mammalian cells, the major player of mitochondrial fission is DRP1 which has a cytoplasmic localization (inactive form) and reversibly associates with OMM at sites of mitochondrial division, where the fission occurs preferentially at ER-contact regions. DRP1 is subject to several post-translational modifications in response to cellular cues. Phosphorylation of DRP1 at Ser637 represents the signal that controls protein translocation from the mitochondria to the cytoplasm. Following mitochondrial depolarization, a rise in cytosolic Ca2+ activates cytosolic calcineurin (CaN) that dephosphorylates DRP1 on Ser637, inducing its translocation to mitochondria. CaN itself is subject to translational regulation by miR-499 in a p53-dependent manner. Conversely, protein kinase A (PKA), after cyclic AMP activation, phosphorylates this residue, leading to the elongation of mitochondria and resistance to various pro-apoptotic insults. DRP1 localization to mitochondria is a process requiring adaptors on the OMM (FIS1, MFF, MiD49, MiD51, and Mcl-1L). The mitochondrial ubiquitin ligase MITOL/MARCH5 promotes mitochondrial division in a DRP1- and MiD49-dependent manner. MITOL/MARCH5 also targets MFN1 and FIS1. The mitochondrial anchored protein ligase (MAPL) induces SUMOylation of DRP1 targeting it to mitochondria and accelerating mitochondrial fission. SUMOylation can be reversed by the SUMO protease SENP5. During mitosis, CDK1/cyclin B phosphorylates DRP1 at Ser616 to induce mitochondrial fission and proper organelle segregation. ERK2 can also phosphorylate DRP1 at Ser616 during MAPK-mediated transformation. Enforced expression of MTP18 correlates with the recruitment of DRP1 to mitochondria. DRP1 can also be S-nitrosylated at Cys644 in response to an increase in NO inducing mitochondrial fission. Finally, an interaction of DRP1 with phosphatidic acid (PA) restrains DRP1 fission activity and shifts the balance toward mitochondrial fusion. OPA1 is involved in inner membrane processing. Alternative splicing of OPA1 gives rise to long forms (L-OPA) that are proteolytically cleaved to short forms (S-OPA) by inner membrane peptidases, OMA1 and YME1L. While L-OPA1 induces mitochondrial fusion and maintains tubular mitochondria, excessive accumulation of S-OPA1 can either induce mitochondrial fusion or accelerate fission and promote mitochondrial fragmentation. The IMM peptidase YME1L constitutively cleaves OPA1 at the S2 site inducing mitochondrial fusion. However, OMA1 cleaves OPA1 at the S1 site constitutively or in response to stress, inducing mitochondrial fragmentation. The expression of OPA1 is under transcriptional control of the NF-κB transcription factor, which governs diverse aspects of cell division and metabolic reprogramming. Post-translational modifications—P: phosphorylation; ub: ubiquitination; SUMO: sumoylation; PA: phosphatidic acid.6,7,10–16
Proteasome-mediated degradation of mitofusins is regulated by ubiquitylation and phosphorylation in response to cellular stimuli. 6 In general, mitofusins are degraded during apoptosis, while decreased proteasomal degradation induces mitochondrial fusion and cell viability (Figure 2). Deletion of either mitofusins or abrogation of GTPase activity prevents mitochondrial fusion, inducing the fragmentation of mitochondria in cultured cells. Moreover, tissue-specific deletion of mitofusins in the central nervous system, heart, muscle, or liver compromises metabolic and organ function, highlighting their importance at both the cellular and organismal levels. 6
Mitochondrial phospholipids have also been shown to be important for mitochondrial membrane fusion. It has been suggested that local alterations of the phospholipid composition on the OMM may induce membrane curvature or destabilize membrane bilayers to promote fusion or fission.6,17 In this regard, phosphatidic acid (PA), a phospholipid that can generate negative curvature of lipid membranes, has an important role in mitochondrial fusion. Phosphatidic acid–preferring phospholipase A1 (PA-PLA1) or Lipin 1 phosphatase decreases PA levels and induces mitochondrial fragmentation. 17 However, mitoPLD, a mitochondrion-localized phospholipase D able to produce PA from cardiolipin, facilitates MFN-mediated fusion and ablation of mitoPLD induced mitochondrial fragmentation in vitro. 6
Inner membrane fusion is mediated by optic atrophy 1 (OPA1), another dynamin-like GTPase anchored to the IMM and mostly exposed to the intermembrane space (IMS). 6 Multiple forms of OPA1 have been described, which are the result of alternative splicing and proteolytic cleavage by mitochondrial proteases that target OPA1 upon dissipation of membrane potential, mitochondrial DNA (mtDNA) loss, or induction of apoptosis. 4 Alternative splicing of OPA1 produces long isoforms (L-OPA1) that are cleaved to short forms (S-OPA1) with different functions on mitochondrial morphology (Figure 2).10,11 The expression of OPA1 is under transcriptional control of the nuclear factor-κB (NF-κB) transcription factor, which governs diverse aspects of cell division and metabolic reprogramming. 18
The inverse process of mitochondrial fragmentation or fission has been observed in response to nutrient excess and cellular dysfunction in cardiovascular and neuromuscular disorders, obesity, and cancer. In normal cells, mitochondrial fission facilitates the autophagic clearance of mitochondria (mitophagy) and allows the adaption of mitochondrial activities to physiological demands. 6 OMM fission is mediated by a cytosolic dynamin family member dynamin-related protein 1 (DRP1) that translocates from the cytosol and binds its receptors on the OMM (fission protein 1 (FIS1), mitochondrial fission factor (MFF), mitochondrial dynamics of 49 kDa protein (MiD49), and mitochondrial dynamics of 51 kDa protein (MiD51)) often at sites where mitochondria make contact with the endoplasmic reticulum. 6 DRP1 forms spirals around mitochondria, and GTP binding and hydrolysis promote a conformational change in DRP1 that results in membrane constriction to sever both IMM and OMM.3,6 DRP1 is regulated post-translationally at different levels including protein phosphorylation, sumoylation, ubiquitination, and S-nitrosylation (Figure 2). 12
Mitochondrial fission is essential for growing and dividing cells to populate them with adequate numbers of mitochondria. Although it is less evident why mitochondrial fission and fusion are also needed for non-proliferating cells, it is probably due to fusion-induced complementation between damaged mitochondria. This allows sharing of RNA, protein components, and lipids with other mitochondria and mitigation of the effects of mtDNA mutations caused by environmental damage. While mitochondrial fusion would prevent mitochondrial damage through complementation, the lack of fusion would favor mitochondrial degradation through autophagy. 3 Thus, non-proliferating neurons cannot survive without mitochondrial fission, and two human diseases, dominant optic atrophy and Charcot–Marie–Tooth disease type 2A, are caused by fusion defects. 4 Mfn1 and Mfn2 knock-out mice die in utero, and although mouse embryo fibroblasts (MEFs) derived from these mice do survive in culture, some of their mitochondria display a reduced mtDNA copy number and decreased membrane potential, causing problems with ATP synthesis. 6 Therefore, although an organism is unable to develop normally without mitochondrial fusion, some cells can survive without this process. These differential requirements of mitochondrial fusion for survival may stem from the distinctive need to distribute mitochondria in large cells, 19 from higher demands on oxidative metabolism in different cell types and developmental stages, or from demands on other functions that are indirectly affected by fusion. 3 In this regard, cancer cells have been proposed to harbor altered mitochondrial metabolism, a phenomenon related to the increased glycolytic activity in cancer cells known as the Warburg effect. It will thus be interesting to understand how dependent are cancer cells on mitochondrial fusion and fission and whether this is particular to one type of cancer.
Mitochondrial shape is controlled by changes in metabolism. In general, mitochondria fuse when they are forced to rely on oxidative phosphorylation by withdrawing glucose as a carbon source, thus maximizing oxidative phosphorylation by stimulating complementation among mitochondria. Fusion is also enhanced by treatments that inhibit protein synthesis, by starvation, and by autophagy induction. Starvation-induced autophagy may enhance fusion by increasing reliance on oxidative phosphorylation through the metabolism of lipids and proteins. 3 More specifically, autophagy induced by starvation or mammalian target of rapamycin (mTOR) inhibition induces increased cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) activation, which phosphorylates DRP1 (at Ser637) and increases its cytoplasmic localization, leading to unopposed mitochondrial fusion. 20 Then, elongated mitochondria are spared from autophagic degradation (mitophagy). Conversely, when elongation is blocked during starvation, mitochondria consume ATP provoking starvation-induced cell death. 20 Mitochondrial fusion is required for mtDNA maintenance, probably because it allows metabolite and mtDNA exchange between mitochondria. Thus, mitochondrial fragmentation due to impaired fusion is often accompanied by bioenergetic defects due to a loss of mtDNA. 21 In agreement with the previous observations linking mitochondrial elongation to cellular survival, stress stimuli leading to protein synthesis inhibition or serum and amino acid deprivation has been shown to produce stress-induced mitochondrial hyperfusion (SIMH), defined as a highly interconnected network that induces increased mitochondrial ATP production and has been proposed to work as an adaptive pro-survival response against stress. 21 Importantly, this mechanism was independent of MFN2, BCL-2-associated X protein (BAX)/BCL-2 homologous antagonist/killer (BAK), and prohibitins but required L-OPA1, MFN1, and the IMM protein stomatin-like protein 2 (SLP-2). 21
Cancer cell metabolism
In cancer, cellular metabolism may be subverted to support tumor growth. For instance, glycolysis is upregulated in almost all cancer cells since it is needed for the synthesis of macromolecules that are required for active proliferation.22,23 This characteristic, known as the Warburg effect, has found clinical utility for imaging of tumors using 18F-fluorodeoxyglucose (18F-FDG) which accumulates in highly glycolytic cancer cells and can be visualized by positron emission tomography. 23 Moreover, this upregulation in glycolysis is currently being explored for cancer therapeutics utilizing the non-metabolizable glucose analog 2-deoxyglucose to block glycolysis and cancer growth. 24 Warburg assumed that “aerobic glycolysis” was a universal property of malignant cells and suggested that cancer is caused by impaired mitochondrial metabolism. However, further studies have challenged this idea, and evidence supporting the idea that dysfunctional mitochondria is the main cause of the Warburg effect is limited. 25 So, although there indeed seems to be a diminished oxidative metabolism in many tumor types, it has been shown that tumor mitochondria do respire and produce ATP. Evidence thus suggests that there is no oxidative phosphorylation to glycolysis switch but rather an upregulation of glycolysis that has also been observed not only in cancer cells but in rapidly proliferating normal cells due to the temporary higher demands in metabolic material and energy for completing the cell proliferation process.23,26
Evidence in support of a decreased oxidative metabolism in cancer cells has been explained in several ways for different tumor types, including decreased pyruvate transporter activity, lower pyruvate oxidation, truncated Krebs cycle, reduced transfer of reducing equivalents to mitochondria, lower mitochondrial content, or defective respiratory chain. 26 Perhaps the most documented cause of a partial inhibition of oxidative phosphorylation in tumor cells is the Crabtree effect or the inhibition by high concentrations of glucose and other hexoses. Also, the acidic pH induced by lactate generation may affect pH-sensitive oxidative enzymes decreasing their function. 26 A general property of transformed cells with dysfunctional mitochondria is increased ROS production. 27 Therefore, it has been suggested that these cells might show a lower threshold of sensitivity to ROS-induced apoptosis. In agreement with this idea, therapies able to specifically target the respiratory chain to further elevate ROS production have been proposed to be useful in cancer cells which show elevated levels of ROS. Indeed, this has been shown with mitochondrial inhibitors like mitocans, As2O3, antimycin A, or rotenone in cancer cells with defective mitochondrial function. 27
However, although there are several examples of tumor cell lines which certainly exhibit a decreased mitochondrial function, that observation does not seem to apply to all tumor cell types. The contribution of glycolysis to the total cellular ATP supply has been estimated to range between 10% and 70% depending on the cell type, 26 indicating that some tumor cell lines have high levels of oxidative phosphorylation and that this pathway might be the predominant energy source. Regarding oncogene-mediated cell transformation, most of the research in cancer cell metabolism has focused on the increased glycolysis in cancer cells. However, recent evidence indicates that some tumor suppressor genes and oncogenes also have an important role in the modulation of mitochondrial function to support carcinogenesis.
Metabolic effects of oncogenes and tumor suppressors
Glycolysis is stimulated in hypoxic regions of the tumor where oxygen levels are low. This occurs by the induction of hypoxia-inducible factor 1 (HIF-1), which is considered a master regulator for the initiation and maintenance of the Warburg effect at the level of gene expression of glycolysis-related enzymes and glucose transporters.22,23,28 HIF-1 induction can be triggered by the accumulation of ROS produced by dysfunctional mitochondria or by Krebs cycle metabolites like succinate that can inhibit HIF-1α prolyl hydroxylases in the cytosol, leading to the stabilization and activation of HIF-1α.22,23 Succinate accumulation in mitochondria can be caused by mutations in succinate dehydrogenase, which has been documented in a familial predisposition to benign tumors. 22 Interestingly, HIF-1 levels are high in certain tumors even under oxygen-rich conditions, indicating that hormones or growth factors might cause stabilization of HIF-1 expression and that it has important roles in tumorigenesis.22,23 Additionally, HIF-1 itself can also function as a negative regulator of mitochondrial biogenesis and oxidative phosphorylation. For example, HIF-1 activation upregulates several mitochondrial proteins including pyruvate dehydrogenase kinase 1, which inhibits the conversion of pyruvate to acetyl-CoA by pyruvate dehydrogenase resulting in decreased mitochondrial respiration.22,29 HIF-1 activation also inhibits MYC transcription and activates its degradation by the proteasome pathway. Since MYC is a positive regulator of PGC-1α and PGC-1β, the mitochondrial coactivators that regulate mitochondrial biogenesis and the expression of the respiratory machinery, its repression by HIF-1 results in the inhibition of mitochondrial biogenesis and oxidative phosphorylation.30,31 Interestingly, the human von Hippel–Lindau (VHL) tumor suppressor (a negative regulator of HIF-1) promoter has a nuclear respiratory factor 1 (NRF-1, a nuclear transcription factor associated with the expression of many genes required for expression and function of the respiratory chain) recognition site. Since NRF-1 is a target for the PGC-1 family coactivators, these proteins may promote mitochondrial oxidative metabolism in part through the upregulation of VHL expression. 30
MYC regulates genes involved in glucose metabolism by binding to and regulating virtually all glycolytic enzyme genes as well as numerous genes involved in mitochondrial function. 23 Deregulated MYC expression is a hallmark of a large fraction of all human tumors acting as an oncogene in approximately 30% of all human cancers. 32 Concerning mitochondria, MYC regulates the expression of over 400 mitochondrial genes, including mitochondrial transcription factor A (TFAM), an mtDNA transcription and maintenance factor, as well as PGC-1α and PGC-1β. 33 Therefore, gain/loss of MYC increases/reduces mitochondrial mass, respectively. In cancer, oncogenic MYC increases cellular biosynthetic and respiratory capacity by upregulating mitochondrial metabolism to support rapid cell proliferation and by regulating the expression of proteins that modulate mitochondrial dynamics.33,34
Other oncogenes commonly activated in cancers affect metabolism, for example, activation of the phosphoinositide 3-kinase (PI3K)-Akt-mTOR pathway can increase cell size, glycolytic activity, metabolism, and cell survival. 22 In this regard, recent data show that mTOR can act as a major regulator of energy production in mitochondria. 35 mTOR regulates mitochondria at multiple levels; transcriptionally, it regulates PGC-1α, ERR-α (a transcription factor downstream of PGC-1α involved in the expression of components of the respiratory chain and control of fatty acid oxidation), ERR-α target genes, and the expression of nuclear-encoded mitochondrial regulators such as TFAM, mitochondrial ribosomal proteins, and components of complex I and V. 35 All these changes result in increased mitochondrial biogenesis and oxidative phosphorylation. Translationally, mTOR regulates mitochondrial metabolism and biogenesis via repression of inhibitory 4E-binding proteins (4E-BPs) that downregulate translation of nuclear-encoded mitochondrial proteins. Finally, mTOR inhibits autophagy, a major catabolic process in the cell that serves the cellular function of eliminating damaged mitochondria.33,35
RAS is an oncogene whose pathway is activated in a variety of tumors, and its activation has been commonly linked to alterations in glycolysis. In this regard, RAS transformation, as well as increased mutant KRas copy number, has been shown to induce metabolic changes in immortalized cells characterized by increased expression of glycolytic enzymes.36,37 Also, RAS transformation has been shown to increase sensitivity to glucose depletion since RAS-transformed cells were unable to upregulate mitochondrial activity after glucose deprivation due to a partially uncoupled mitochondrial respiration. 36 In contrast, increased mitochondrial activity has been linked to resistance to therapy and recurrence in RAS-transformed cancers probably due to upregulation of PGC-1α. 33 In BRAF or NRAS mutant melanomas, resistance to mitogen-activated protein kinase (MAPK) inhibitors (which are activated by RAS transformation) was partially due to a switch to oxidative metabolism mediated by PGC-1α upregulation and was overcome by mTOR inhibition which repressed PGC-1α expression. 33 Also, in a mouse model of KRas-mutant pancreatic ductal adenocarcinoma (PDAC), cells that survive oncogene ablation showed increased PGC-1α expression and mitochondrial function. Interestingly, the reliance on mitochondrial respiration resulted in sensitivity to oxidative phosphorylation inhibitors. 38
Therefore, oncogenes commonly activated in cancer not only activate glycolysis as Warburg initially proposed but can also activate mitochondrial metabolism. Whether this mitochondrial activation is particular to certain cells within a tumor (those that are not in hypoxic regions where HIF-1 is stimulated or to those cells which are resistant to therapy) or whether this is a metabolic effect in tumor cells with certain oncogenic mutations of specific cancer types or tissues remains to be determined.
Of special interest to cancer cell metabolism is the fact that p53, a crucial tumor suppressor, long recognized to suppress cancer through the induction of cell-cycle-arrest or apoptosis programs in response to a plethora of different cellular stress signals, has been closely related to tumor cell metabolism. These functions include the regulation of metabolism, autophagy, and the oxidative status of the cell. 28 Concerning tumor cell metabolism, p53 can modulate the balance between glycolysis and oxidative phosphorylation. It can induce the expression of the protein synthesis of cytochrome c oxidase 2 (SCO2) that is required for the assembly of cytochrome c oxidase (COX). 22 p53 mutation in tumors causes downregulation of mitochondrial respiration as a result of COX deficiency and a shift of cellular metabolism toward glycolysis. Another target of p53 is TP53-induced glycolysis and apoptosis regulator (TIGAR), which decreases fructose-2,6-bisphospate levels in cells, resulting in the inhibition of glycolysis, while stimulating nicotinamide adenine dinucleotide phosphate (NADPH) generation through the pentose-phosphate shunt. 22 These studies suggest that p53 is an important regulator of tumor cell metabolism and that cells with wild-type p53 and cells with mutations or deletions in this gene probably have differences in glycolytic and mitochondrial metabolism. Importantly, more than half of human cancers of a wide variety of types harbor p53 mutations and even more have alterations in the pathways that regulate p53 stability within the cell. 28 Importantly, p53 has an important relationship with HIF-1, and the amplification of HIF-1-dependent responses to hypoxia via the loss of p53 function contributes to the angiogenic switch, promoting cancer progression. 39 Since HIF-1 can downregulate mitochondrial activity; those tumors with HIF-1 activation and p53 mutations or deletions could be the ones showing a dramatic increase in glycolysis as well as an impaired mitochondrial metabolism. In other tumors, the series of mutations driving tumorigenesis could have a specific effect on tumor cell metabolism.
Mitochondrial dynamics and cell death
Apoptosis is triggered in response to various types of stress that cancer cells experience during tumorigenesis such as high levels of oncogene signaling, DNA damage associated with hyper proliferation, or anti-cancer therapy. Also, apoptosis has been shown to be attenuated in tumors, and a resistance to cell death has been listed as one of the hallmarks of cancer cells. 40 Mitochondria are a focal point of apoptotic signaling. Diverse death stimuli ranging from DNA damage to metabolic stress due to immune cell-mediated receptor ligation converge on mitochondria to trigger the release of cytochrome c and other death-promoting proteins from the IMS. The release of cytochrome c is considered a critical decision point in apoptosis and occurs through mitochondrial outer membrane permeabilization (MOMP), which is regulated by the B-cell lymphoma 2 (BCL2) protein family members, 41 and alterations in the intrinsic or mitochondrial apoptotic pathway have been implicated as a barrier to anti-cancer therapy. 40
The BCL2 family of proteins is divided into pro- and anti-apoptotic members, and they all share combinations of BCL2 homology (BH) domains. BCL2-like proteins (BCL-XL, BCL-W, A1, and MCL-1) are pro-survival and antagonize the pro-apoptotic members of the family (BAX, BAK, and BCL2 related ovarian killer (BOK)), which mediate mitochondrial apoptosis through permeabilization of the OMM, allowing efflux of apoptogenic proteins. 42 BH3-only proteins (BCL2-like 11 (BIM), BCL2 associated agonist of cell death (BAD), BH3-interacting domain death agonist (BID), p53 upregulated modulator of apoptosis (PUMA), and NOXA) of the BCL2 family share this BH3 interaction domain and antagonize BCL2 like proteins. The regulation of interactions between pro-apoptotic and anti-apoptotic members of the BCL2 family of proteins determines whether a cell lives or dies in response to a stimulus. 42 Once in the cytosol, cytochrome c induces the oligomerization of caspase-9, apoptotic protease activating factor 1 (APAF-1), and ATP to form the apoptosome that goes on to activate the executioner caspases, caspase-3 and caspase-7. These terminal caspases cleave various cellular substrates that lead to cellular dismantling through apoptosis.41–43 Interestingly, highly glycolytic cells like neurons and cancer cells can be refractory to cytochrome c–induced caspase activation. In these cells, glucose-stimulated production of glutathione as a result of NADPH production through the pentose-phosphate pathway inactivated cytochrome c by keeping it in its reduced state. 41
The BCL2 family proteins also have important effects on metabolism. For instance, phosphorylated BAD can promote hepatic glycolysis by activating glucokinase in response to the fed state. 44 Also, proteins from the BCL2 family have been shown to modulate the balance between mitochondrial fission and fusion. In this regard, mitochondrial fission has been generally associated with apoptotic cell death. In healthy cells (i.e. in the absence of apoptotic stimuli), the combined deletion of BAK and BAX results in reduced fusion. In this setting, non-activated BAX may promote fusion by altering MFN2 distribution, mobility, and/or complex assembly.43,45 Also, SIMH, which leads to highly elongated mitochondria, has been shown to delay BAX activation upon exposure of cells to apoptotic stimuli. 43
During apoptosis, mitochondrial fission occurs as an early event, occurring within the same time frame as activation of BAX and MOMP. In this regard, diverse interactions have been described for DRP1 and pro-apoptotic BCL2 family proteins. In apoptotic cells with activated BAX (recruited to the mitochondrial membrane), DRP1, which normally cycles between the cytosol and the mitochondrial membrane, stably associates with the OMM due to BAX/BAK dependent SUMOylation and precedes MOMP. 46 Also, DRP1 binds and is required for BAX mitochondrial translocation after an apoptotic stimulus. 47 The constricting ability of DRP1 may contribute to the mitochondrial membrane curvature required for the efficient insertion of BAX and mitochondrial cristae remodeling, suggesting a pro-apoptotic role for DRP1. Interference with DRP1 function during apoptosis results in a block of mitochondrial fission, leading to long interconnected organelles as well as a delay in cell death. 46 In this study, the authors suggest that during apoptosis, cycling of DRP1 between the cytoplasm and the mitochondria mediates mitochondrial fission, whereas BAX/BAK dependent membrane-associated DRP1 is involved in the subsequent apoptotic events, such as the remodeling of the mitochondrial membrane that leads to MOMP and a loss of membrane potential. Interestingly, DRP1 functions in apoptosis could be independent of or complementary to mitochondrial fission, since DRP1-induced mitochondrial fragmentation by itself is not sufficient to induce apoptosis.46,47
It has also been suggested that hyperfragmented mitochondria are not involved in apoptosis and that MFN1 establishes a mitochondrial size (fragmented, but not hyperfragmented) that is permissive for pro-apoptotic BCL2 family function. 48 In this study, cells with hyperfragmented mitochondria (less than 0.5 µm) failed to support BAX-dependent membrane association and permeabilization due to an inability to stabilize BAX–membrane interactions. Bioenergetics of large and small mitochondria were similar but only larger mitochondria supported BAX integration to the OMM and mitochondrial pore formation. The authors suggest that while mitochondrial hyperfusion can protect from cell death, a balanced, non–hyperfragmented mitochondrial network is required for BAX-dependent MOMP after terminal ER stress or chemotherapeutic drug treatment. In agreement with the previous findings, increased sensitization to cisplatin- or paclitaxel-induced apoptosis was observed after DRP1 inhibition with mitochondrial division inhibitor-1 (mDIVI-1), which induced mitochondrial fusion in the human melanoma cell line A375, which normally displays hyperfragmented mitochondria. 48
Mitochondrial dynamics and cell cycle progression
Mitochondria grow continuously throughout the cell cycle and their dynamics and organization are tightly controlled across its different phases to ensure proper cell division. 49 In mammalian cells, evidence suggests that as cells progress through G1, mitochondrial membrane potential and respiration markedly increase. 50 Then, at G1/S, mitochondria undergo a marked alteration in morphology, forming a giant and tubular network with hyperpolarized and highly coupled mitochondria.49,50 This network is associated with increased energy production to prepare for DNA synthesis. At this stage, mitochondrial hyperfusion also facilitates the mixing of mitochondrial contents to promote their homogeneity within the cell, ensuring proper mitochondrial inheritance following cellular division. Mitochondrial hyperfusion triggers S-phase initiation and this alone is sufficient to drive G0 quiescent cells into S phase of the cell cycle. 49
During the following S, G2, and M phases, mitochondria become increasingly fragmented, reaching the highest fragmentation at mitosis to assure equal segregation of mitochondrial contents between daughter cells. 49 Mitotic mitochondrial fission is directly regulated by kinases involved in mitosis, like Aurora A kinase that promotes RalA relocalization to mitochondrial membranes and recruits the effector RalBP1. RalBP1 functions as a scaffold for cyclin B/cyclin-dependent kinase 1 (cdk1) that promotes phosphorylation of DRP1 at Ser616 resulting in mitochondrial fission. Sustained mitochondrial hyperfusion beyond the G1/S border induces replication stress causing ATM-dependent G2/M delay and chromosomal instability during mitosis. Also, prolonged mitochondrial fusion causes mitochondrial bridges between daughter cells resulting in a defective cytokinesis, unequal distribution of mitochondria, incorrect segregation of chromosomes, and aneuploidy. Cells that experience persistent mitochondrial hyperfusion during mitosis present genomic instability characterized by the presence of micronuclei and disorganized nuclear structure. 49
Mitochondrial dynamics and cancer
Cancer is a disease characterized by inappropriate cell proliferation, dysregulated cell cycle control, and defects in apoptosis. Since mitochondrial dynamics has been shown to be involved in the aforementioned mechanisms, it is not surprising to find recent evidence that strongly relate the processes of mitochondrial fusion and fission to cancer progression in diverse types of cancer.
As in normal cells, mitochondrial dynamics in cancer cells have important implications for cellular function as well as for sustaining proliferation and resistance to diverse types of stress. In general, the majority of studies that have explored mitochondrial morphology in tumor cells support a pro-tumorigenic role for mitochondrial fission (Table 1). 51 Reduced cancer cell growth and/or spontaneous apoptosis induced by DRP1 inhibition alone or in combination with chemotherapy have been observed both in vitro and in vivo in several cancer types, including breast, colon, lung, pancreatic, hepatocellular carcinoma, and melanoma.49,52–58 Moreover, alterations in the levels of proteins regulating mitochondrial dynamics have been suggested as potential biomarkers of malignancy or as a target for therapy in diverse tumor types (Table 2). In this regard, inhibition of mitochondrial fragmentation by silencing of DRP1 has been associated with increased genomic instability 49 and reduced migration and invasion 59 in breast cancer cell lines which normally have a high level of fragmented mitochondria. The mechanism underlying cellular dysfunction and cell death induced by decreased mitochondrial fission seems to be the replicative stress and mitotic defects that severely affect the genomic integrity of cancer cells. Impaired mitochondrial fission also causes mitochondrial dysfunction characterized by accumulation of mtDNA mutations and generation of ROS. This combined effect of genome instability and mitochondrial dysfunction may eventually result in the activation of the mitochondrial apoptotic pathway, suggesting that inhibition of mitochondrial fission could be a potential mechanism to enhance cancer cell apoptosis, to increase sensitivity to therapy, or to potentially circumvent resistance of cancer cells to conventional chemotherapy.49,60
Mitochondrial dynamics and its relationship with cancer-related processes.

UV: ultraviolet; TIC: tumor-initiating cells; OMM: outer mitochondrial membrane.
Some of the recent studies regarding mitochondrial fusion or fission and the suggested role in different aspects of apoptosis or cancer progression are mentioned.
Alterations in proteins regulating mitochondrial dynamics in tumor samples and their suggested use as prognostic or diagnostic biomarkers.

MAPK: mitogen-activated protein kinase; ERK: extracellular signal–regulated protein kinase; mRNA: messenger RNA.
In agreement with the previous observations, it has also been reported that several cancer cell lines or cells that have been transformed with oncogenes have more fragmented mitochondria than their non-transformed controls.48,55,56,59 At least one of the molecular mechanisms involved in the maintenance of fragmented mitochondria in cancer cells has been recently elucidated and it has been proposed to involve the MAPK signaling pathway, a proliferation pathway activated in a variety of tumors. In normal cells, the RAS-RAF-MEK-ERK (MAPK) pathway is activated by growth factors and hormone signaling and its precise regulation controls proliferation and cell death. In cancer cells, excess signaling by growth factors, hormones, or oncogenic mutations (like RASG12V or BRAFV600E) activate this pathway which results in aberrant signaling, uncontrolled proliferation, and silencing of the cell death machinery. A hallmark feature of cancer cells with MAPK oncogenic activation is an increase in anaerobic glycolysis (Warburg effect) and glutaminolysis. More recently, mitochondrial defects characterized by mitochondrial fragmentation with decreased mitochondrial oxygen consumption rate and mitochondrial ATP production have been described in RAS-transformed cells. 55 While the RalGEF pathway has been shown to promote mitochondrial fission during mitosis through DRP1 phosphorylation by Cdk1, RAS-transformed cells and tumors show mitochondrial fragmentation independent of the cell cycle, and this constitutive mitochondrial fission has been attributed to the activation of ERK2.55,56 DRP1 has been shown to be phosphorylated by ERK2 on Ser616, and this phosphorylation is required for mitochondrial fission and tumor growth in RAS-transformed tumors, emphasizing the need for MAPK activation and consequent mitochondrial fragmentation in tumors expressing oncogenic RAS. 56 Interestingly, RAS is mutated in up to 30% of all tumors and in 90% of PDACs, and BRAFV600E mutations have been identified in approximately 66% of melanomas and in a smaller percentage in other tumors (thyroid, colon, ovarian cancer, and sarcomas). 61 Tumors or cell lines from these types of cancer are known to have an altered metabolism and increased mitochondrial fragmentation55,56. Besides, DRP1 knockdown, expression of the dominant-negative DRP1-K38A or inhibition of DRP1 phosphorylation on Ser616 decreased tumor growth in a RAS-driven mouse tumor model, suggesting that DRP1 is a potential therapeutic target in tumors expressing oncogenic RAS. Also, DRP1S616 phosphorylation was found to correlate with BRAFV600E in human melanoma samples, 55 and DRP1 phosphorylation or RAS transformation led to decreased mitochondrial membrane potential, increased production of mitochondrial ROS, and reduced basal and maximal oxygen consumption rates, 55 indicating that ERK2-mediated DRP1 phosphorylation is responsible for decreased mitochondrial function in RAS-transformed cells. In mouse tumors, DRP1 inhibition induced a decrease in tumor vasculature and decreased vascular endothelial growth factor (VEGF) messenger RNA (mRNA). 56 The authors suggest that one of the possible mechanisms of tumorigenesis induced by MAPK-mediated mitochondrial fission could be the activation of proangiogenic signaling pathways, which are sensitive to mitochondria-derived ROS. Also, with regard to MAPK-induced mitochondrial fragmentation, c-Jun N-terminal protein kinase (JNK) activation during chemotherapy-induced apoptosis has been related to proteasomal degradation of MFN2 through the activation of Huwe1 ubiquitin ligase, decreasing mitochondrial fusion and inducing apoptosis. 13 Therefore, MAPK activation in cancer has been linked to mitochondrial fission and can induce transformation in some cases (RAS transformation) and apoptosis in other contexts (chemotherapy-induced apoptosis). Importantly, in the first case, DRP1 inhibition (inhibition of fission) decreased tumor growth in a mouse model and in patient-derived pancreatic cancer cell lines; 56 while in the second case, MFN2 knockdown (inhibition of fusion) increased apoptosis in doxorubicin-treated cells, highlighting the need to precisely understand how to manipulate mitochondrial dynamics for cancer therapy.
MYC is another oncogene whose effects seem to be mediated by the regulation of mitochondrial dynamics, and in contrast to RAS, MYC has been suggested to induce mitochondrial fusion.32,34 Graves et al. 34 reported augmented mitochondrial mass with increased membrane polarization, a correction of OXPHOS deficiency, and mitochondrial fusion upon Myc re-expression in myc−/− fibroblasts. Myc re-expression altered the levels of proteins involved in mitochondrial dynamics ultimately favoring fusion and increasing mitochondrial mass. 34 Also, in a recent study, MYC induced PLD6 (a phospholipase of the OMM) expression and mitochondrial fusion to promote biosynthesis of metabolic precursors and MYC-driven cell growth. 32 Since MYC potently stimulates cell growth and proliferation, the authors suggest that MYC-induced mitochondrial fusion is responsible for increased lipid, nucleotide, and protein metabolism, which strain cellular energetic resources and hence activate adenosine monophosphate–activated protein kinase (AMPK, which is activated in response to high AMP and low ATP levels) to inhibit yes-associated protein (YAP)/TAZ coactivator activity. In this work, induction of mitochondrial fusion by mDIVI-1 was sufficient to activate AMPK indicating that MYC exerts at least some of its effects on metabolism and cell proliferation by inducing mitochondrial fusion. 32 Interestingly, MYC has been found to be highly expressed in triple-negative breast tumors and has been associated with poor prognosis in breast cancer patients, 32 suggesting a potential use for inhibitors of mitochondrial fusion for this type of cancer.
Regarding metastasis, human breast carcinomas and metastasis to lymph nodes have been found to have higher levels of DRP1 than ductal carcinoma in situ samples or than the normal/adjacent tissue. 59 Also, invasive breast cancer cell lines have been found to have fragmented mitochondria, higher DRP1 and DRP1S616 levels, as well as lower MFN1 than a non-metastatic cell line. 59 In the same study, DRP1 knockdown, DRP1 inhibition with mDIVI-1, or MFN-1/2 expression decreased migration and invasion in the metastatic cell lines without affecting cell cycle or viability. Mitochondrial fission induced cell spreading and increased lamellipodia formation as well as mitochondrial distribution to the lamellipodia region at the leading edge of invading cells in response to chemoattractants, suggesting a crucial role of mitochondrial fission in the migration and invasion of breast cancer cells. 59 Also, since hypoxia is a major stimulator of migration and invasion in cancer cells, another study found that hypoxia increased the expression of DRP1 and mitochondrial fission in a glioblastoma cell line. In the same study, hypoxia-induced migration was decreased with mDIVI-1 treatment or a dominant-negative DRP1-K38A. 62 These studies suggest that targeting mitochondrial fission is likely to have a therapeutic benefit in highly invasive cancers like glioblastoma, which is one of the most aggressive cerebral gliomas, or in other highly metastatic cancers.
Other studies have found that tumor cells with increased mitochondrial fragmentation are more sensitive to metabolic stress and have proposed mitochondrial inhibition as a therapeutic target in these types of cancers. For example, in prostate cancer, androgens and androgen receptors play critical roles in the proliferation of prostate cancer through transcriptional regulation of target genes. Androgens have been found to upregulate the expression of DRP1, facilitating mitochondrial fragmentation and apoptosis in prostate cancer cell lines in response to mitochondrial stress. Interestingly, in the same study, clinical tissue samples of prostate carcinoma showed high levels of DRP1 in androgen-dependent cancers but not in androgen independent cancers. The authors suggest that androgens might function as a pro-apoptotic stimulus when combined with agents that affect mitochondrial function in androgen-dependent cancers. 63 Also, with regard to hormonal regulation of mitochondrial dynamics, estradiol treatment has been shown to affect the levels of proteins regulating mitochondrial dynamics and to induce mitochondrial fusion in the MCF-7, estrogen receptor positive cell line. 64 The above-mentioned studies indicate that hormones have a strong influence on mitochondrial dynamics and the overall effect might involve hormone-specific effects, the type of malignancy, or the type of hormone receptors present in the cancer cell or tumor tissue, 65 underscoring the importance of understanding how to manipulate mitochondrial dynamics for the treatment of hormone-related malignancies. Besides decreasing mitochondrial metabolism, it has also been shown that mitochondrial fragmentation increases glycolytic metabolism and makes cancer cells more sensitive to glycolysis inhibitors. 66 In this study, survivin, a member of the Inhibitor of Apoptosis Protein family which is overexpressed in neuroblastoma, induced increased DRP1 levels and mitochondrial fragmentation in a neuroblastoma cell line. Mitochondrial fission increased glycolysis and lactate production and made cells more sensitive to 2-deoxyglucose alone or in combination with chemotherapy. These studies suggest that mitochondrial fragmentation could be a biomarker for treatment with either or both mitochondrial and glycolytic metabolic inhibitors.
It has also been suggested that mitochondrial inhibition could be a potential therapeutic target for the highly tumorigenic tumor-initiating cells (TICs). TICs or cancer stem cells have been defined as cells within the tumor that possess clonal long-term repopulation and self-renewal capacity. These cells have been proposed to be responsible for therapy failure and the formation of metastasis.67,68 In a PDAC model, dormant tumor cells surviving oncogene-ablation (KRAS withdrawal) were responsible for tumor relapse and had TIC features. These TICs showed prominent expression of genes governing electron transport chain, autophagy, lysosome activity, mitochondrial and peroxisomal oxidation, increased PGC-1-α and mitochondrial mass, increased oxygen consumption rate as well as a strong reliance on mitochondrial respiration for survival, and a decreased dependence on glycolysis for cellular energetics. 38 In the same study, mitochondria from TICs had an altered morphology, were hyperpolarized, and produced more ROS. The authors suggest that altered metabolic and mitochondrial functions are critical features of TICs in this model, making them more resistant to apoptosis. Interestingly, OXPHOS inhibition by oligomycin decreased ATP content and induced cell death in TICs, which were unable to upregulate glycolysis when compared to KRAS expressing cells from the same model. Altered metabolism in these TICs was attributed to increased protein and fatty acid metabolism, making them more sensitive to autophagy (which mediates lipid droplet degradation) and β-oxidation inhibitors. These findings indicate that TICs are more dependent on mitochondrial respiration than the highly glycolytic bulk of tumor cells. 38
In agreement with the previous report, in a different model, brain TICs isolated from tumor xenografts or from primary tumors have been found to have fragmented mitochondria and increased levels of activating DRP1S616 phosphorylation relative to non-TICs, which showed higher levels of DRP1S637 inactivating phosphorylation. Interestingly, transfection with a phosphomimetic, inactivation-incompetent double-mutant DRP1S616E, S637A induced expression of stem cell markers in non-TIC tumor cells, and DRP1 knockdown or its pharmacological inhibition with mDIVI-1 decreased tumor growth in two brain tumor mouse models. Furthermore, while total DRP1 levels were similar in normal and neoplastic brain tissues, activating phosphorylation of DRP1 (DRP1S616) was increased in glioblastomas, and a strong inverse correlation was found between DRP1S616 and poor survival in glioblastoma patients. 69 In this work, the authors suggest that cyclin-dependent kinase 5 (CDK5) was responsible for DRP1 activation in brain TICs and that calcium-/calmodulin-dependent protein kinase type 2 (CAMK2) was responsible for inhibitory phosphorylation of DRP1 in Ser637 in non-TICs. 69 Since glioblastomas rank among the most lethal cancers, it will be interesting to explore DRP1 inhibition as a therapeutic strategy for these tumors.
In addition, mammospheres of breast cancer cell lines, which are known to enrich TICs, as well as breast cancer tumors, overexpress mitochondrial enzymes, proteins involved in mitochondrial biogenesis, and protein inhibitors of mitophagy when compared to their surrounding tissue. 70 The previous findings suggest that tumor cells, and more specifically TICs, resist stress by increasing their ATP production through oxidative mitochondrial metabolism. Regarding mitochondrial dynamics, MYC-induced proliferation was shown to be mediated by the induction of PLD6-mediated mitochondrial fusion and resulted in a decrease in mammosphere forming ability in CD44high/CD24low mammary epithelial cells. 32 This work, together with the previously mentioned studies, suggests a relationship between mitochondrial fragmentation and TIC features or between mitochondrial fusion and a decrease in TIC characteristics.
Other studies suggest that mitochondrial fusion could be involved in resistance to therapy. Cervical and ovarian cancer cell lines resistant to cisplatin showed a higher proportion of tubular mitochondria and reduced sensitivity to cisplatin-induced mitochondrial fragmentation and apoptosis induction than their non-resistant controls. In this work, the authors show that p53 re-expression induces mitochondrial fragmentation in p53 mut or p53 null cell lines. The mechanism by which p53 regulates apoptosis in cisplatin-sensitive cells involves cisplatin-mediated p53 phosphorylation which translocates to the mitochondria and binds prohibitin1, releasing OPA1 and allowing its processing to S-OPA1 by Oma1, whose proteolytic activation is also induced by p53, inducing mitochondrial fragmentation and apoptosis. 10 This will be an interesting hypothesis since, as we mentioned previously, more than half of human cancers harbor p53 mutations and even more have alterations in the pathways that regulate its stability within the cell, 28 suggesting that the induction of mitochondrial fragmentation could help restore the sensibility to therapy.
Other links have been described between the mitochondrial fusion–fission machinery and p53. p53 has been shown to bind DRP1, which mediates the monoubiquitination of p53 by mouse double minute 2 homolog (MDM2). This allows p53 to translocate to the mitochondria and initiate necrosis in response to oxidative stress. Also, p53 directly promotes DRP1 expression in conditions of oxidative stress, which induces mitochondrial fission and the activation of caspase-3, and the enforced expression of miR-30 attenuated mitochondrial fission by repressing p53. 71 Since p53 also transcriptionally regulates the expression of proteins that promote mitochondrial apoptosis, it will be interesting to determine whether its role in the induction of mitochondrial fragmentation causes cell death by itself or if it is only a mechanism to facilitate cell dismissal during apoptosis.
Discussion
Since changes in mitochondrial morphology are closely linked to important cellular functions, including those altered in cancer, it is not surprising that alterations in mitochondrial dynamics are associated with tumor progression or resistance to therapy. Several alterations in mitochondrial mass and ultrastructure have been reported in neoplastic tissues, including ultrastructural heterogeneity, cristal disorganization, altered mitochondrial membranes, matricial vacuoles, mitochondrial swelling, and distorted shapes, 72 suggesting alterations in function when compared to normal cells, but the functional implications of these observations or their importance for cancer treatment are not clear. Moreover, despite an increased understanding of the molecular mechanisms regulating mitochondrial fusion and fission and their role in certain cancer types or cancer-related situations, a precise understanding of a mechanism to manipulate mitochondrial dynamics is not being used for cancer therapy.
The main complication for effectively targeting mitochondrial fusion or fission for therapy or for using mitochondrial morphology as a cancer biomarker is the context-dependent role of mitochondria in diverse types of cancer or settings (Table 1). However, with a few exceptions, most of the evidence suggests that increased mitochondrial fragmentation can be used as a diagnostic biomarker for tumor progression or for certain oncogene mutations (Table 2), although whether this is a general feature for all tumor types remains to be determined.
Oncogene transformation has been shown to have diverse effects on mitochondrial morphologies: while RAS transformation was shown to mediate mitochondrial fragmentation,55,56 MYC-induced oncogenesis induced mitochondrial elongation.32,34 Importantly, these changes in mitochondrial dynamics were shown to be relevant for oncogene-mediated transformation and proliferation. Besides, p53, the most frequently mutated tumor suppressor gene, induced mitochondrial fragmentation and apoptosis when re-expressed in p53-mutated or null ovarian cancer cell lines. 10 The aforementioned evidence suggests that the array of mutations in oncogenes or tumor suppressors could define mitochondrial morphology and probably mitochondrial function within a particular tumor. In this regard, steroid hormones were also shown to affect mitochondrial dynamics in hormone-related malignancies: while androgens increased mitochondrial fragmentation in androgen-sensitive prostate cancer, estradiol induced mitochondrial fusion in an estrogen receptor–positive breast cancer cell line. Thus, it will be important to identify whether changes in mitochondrial morphology are specific to an array of cancer mutations or to cancer cells in a tissue, whether these changes contribute to tumor progression, and whether mitochondrial dynamics can be effectively targeted for therapy.
Regarding treatment, the induction of mitochondrial fragmentation is generally associated with a pro-apoptotic effect in cancer cells treated with chemotherapy or ultraviolet (UV) radiation, suggesting that the induction of fission or inhibition of fusion could be potentially used in combination with other therapies during cancer treatment in several malignancies. However, the proposal that mitochondria need a minimal size permissive for apoptosis and that hyperfragmented mitochondria are not involved in apoptosis 48 raises important questions as to how to induce mitochondrial fragmentation and to what extent. Importantly, in cancer cells that normally display fragmented mitochondria, DRP1 inhibition sensitized them to treatment10,48 or induced cell death per se, 49 underscoring the importance of understanding whether the induction of mitochondrial fragmentation or inhibition of fusion will only be useful to target cancer cells with tubular mitochondria.
The suggestion that TICs, those cells within a tumor that have tumor-initiating capacity and that have been suggested to be responsible for tumor relapse, display increased mitochondrial fragmentation and are more sensitive to mitochondrial stress in several tissues (i.e. a pancreatic cancer model, 38 breast cancer mammospheres, 70 or brain tumor TICs from tumor xenografts or primary tumors 69 ) raises an important suggestion for targeting mitochondria in combination with cancer therapy in order to eliminate TICs in the tumor. Importantly, some of the above-mentioned studies suggest targeting mitochondrial oxidative metabolism,38,70 and regarding mitochondrial dynamics, Xie et al. 69 showed that DRP1 knockdown or mDIVI-1 treatment decreased brain TICs. Perhaps one of the most important findings of this study is the fact that expression of DRP1 with two mutations (Ser616E that mimics an activating phosphorylation and Ser637A that blocks inhibitory phosphorylation) induced mitochondrial fragmentation and the expression of some core stem cell regulators and repression of differentiation markers in non-TICs. Therefore, mitochondrial fragmentation by itself can induce TIC features in cancer, making mitochondrial fission a potential target in these malignancies. Whether this is particular for brain TICs or whether it is a general feature of TICs in other tissues remains to be established.
Finally, several questions remain to be answered. Are there four mitochondrial states (hyperfused, fused, fragmented, and hyperfragmented) rather than just two (fused and fragmented) and do they have different functions? Are there particular biomarkers for each of these morphologies? How is this relevant to the different cancer cells? Is mitochondrial fission only a feature of MAPK-transformed cancers and fusion a feature of MYC transformation? How do other oncogenes affect mitochondrial dynamics? How does mitochondrial dynamics manipulation combine with conventional chemotherapies used to treat these types of cancer? Is mitochondrial inhibition useful for TICs in all cancer types? Is mitochondrial fission necessary for migration in all cancer cells? It is very likely that a more precise understanding of the mechanisms governing mitochondrial dynamics and their role in cancer will be available soon, allowing for an improvement in cancer-related therapies as well as for their use as cancer-related biomarkers.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the following project from the Instituto Mexicano del Seguro Social (IMSS): CTFIS/1ORD/012/2011. P.M. is a recipient of a Cátedra CONACYT (Consejo Nacional de Ciencia y Tecnología) for young investigators (Cátedras CONACYT/3103).