
Abstract
Emerging evidence indicates that microRNAs play critical roles in carcinogenesis and cancer progression. In this study, miR-133a was found to be significantly downregulated in colon tumor tissues. We aimed to determine its biological function, molecular mechanisms, and direct target genes in colorectal cancer. From these results, we found that miR-133a was significantly downregulated in primary tumor tissues and colon cancer cell lines. Ectopic expression of miR-133a in colon cancer cell lines significantly suppressed cell growth, as evidenced by cell viability and colony formation assays, as well as reduced xenograft tumor growth in nude mice. However, the effect of miR-133a was abolished by the overexpression of eIF4A1. Moreover, miR-133a inhibited cellular migration and invasiveness. A luciferase activity assay revealed oncogene eukaryotic translation initiation factor 4A1 as a direct target gene of miR-133a, whose expression was inversely correlated with that of miR-133a. Our results demonstrate that miR-133a plays a pivotal role in colorectal cancer by inhibiting cell proliferation, invasion, and migration by targeting oncogenic eukaryotic translation initiation factor 4A1, which acts as a tumor suppressor and may provide a new potential therapeutic target in colorectal cancer.
Introduction
Colorectal cancer (CRC) is one of the most malignant diseases and the second leading cause of tumor-related deaths in Western countries. 1 The incidence of CRC is rapidly rising in Asian countries, which were formerly considered as low-risk areas. 2 Despite previous studies on the subject, there is a growing need to identify the underlying molecular pathogenesis of CRC. Over the last decade, microRNAs (miRNAs) have emerged as key players in carcinogenesis. 3 The aberrant expression of miRNAs has been demonstrated to play a critical role in the initiation and progression of several human cancers through post-transcriptional regulation of gene expression. 4
miRNAs, which are typically 18–22 nucleotides in length, are small noncoding RNAs that negatively regulate gene expression through base pairing with the 3′-untranslated regions (UTRs) of target messenger RNAs (mRNAs), causing mRNA degradation and/or translational repression. 5 miRNAs have also been shown to play a key role in the development of CRC. 6 miRNAs in CRC exhibit oncogenic or tumor suppressive activity by directly regulating oncogenes or tumor suppressor genes. miR-133a has been confirmed as a tumor suppressor in gastric cancer, lung cancer, breast cancer, and prostate cancer by suppressing cellular proliferation, migration, and invasion.7–9 However, the functional role and mechanistic action of miR-133a in CRC remain largely unclear. In this study, we aimed to determine its biological function, molecular basis, and target genes in CRC.
Materials and methods
Cells and surgical specimens
Three human colon cancer cell lines (HCT116, HT29, and SW480) and the human benign colon cells NCM460 were purchased from Shanghai Institutes for Biological Sciences (Shanghai, China). All cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA), 100 mg/mL streptomycin, and 100 U/mL penicillin in a humidified incubator containing 5% CO2 at 37°C. Specimens of primary CRC tissues and matched adjacent noncancerous tissues (NTs) were collected and paired from 51 patients with CRC between October 2007 and June 2011 following surgical resection at The First Affiliated Hospital of Wenzhou Medical University.
RNA isolation and quantitative reverse transcription polymerase chain reaction assay
For eukaryotic translation initiation factor 4A1 (eIF4A1) detection, total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Total RNA (2 µL) was reverse transcribed in a 20-µL reaction system using a Quant Reverse Transcriptase kit (TianGen Biotech Co., Ltd, Beijing, China). Real-time quantitative polymerase chain reaction (PCR) analysis was performed using a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). RT-primers of eIF4A1 and miR-133a mRNAs were designed and synthesized by GenePharma (Shanghai, China) as follows: eIF4A1, 5′-AAG GCG TCA TCG AGA GTA ACT-3′ (forward) and 5′-ATG TGG CCG TTT TCC CAG TC-3′ (reverse); glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′-AGC CTT CTC CAT GGT GGT GAA-3′ (forward) and 5′-ATC ACC ATC TTC CAG GAG CGA-3′ (reverse); miR-133a, 5′-ATA AGA ATG CGG CCG CAT TCC AAA CTA GCA GCA CTA-3′ (forward) and 5′-AGC TTT GTT TAA ACT TAA CCA TTC TAG CTT TTC C-3′ (reverse); and U6, 5′-CTC GCT TCG GCA GCA CA-3′ (forward) and 5′-AAC GCT TCA CGA ATT TGC GT-3′. The relative quantification of eIF4A1 mRNA was normalized using GAPDH, and the relative quantification of miR-133a was normalized using U6. The PCR program was set as follows: 95°C for 10 min, followed by 35 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 45 s.
miRNA transfection and RNA interference
Human miR-133a precursor (pre-miR-133a, MC10413) and the miRNA mimic negative control (control) were purchased from Ambion, Life Technologies (Austin, TX, USA). Cells were seeded on 24-well plates at 60% confluence and transfected with 15 pmol miRNA per well using Lipofectamine 2000 (Invitrogen). Small interfering RNAs (siRNAs) against human eIF4A1 (TG313247, OriGene, Rockville, MD, USA) were delivered into cells using Lipofectamine 2000. Cells transfected with miRNA or siRNA were harvested 12 to 48 h post-transfection.
Cell viability assay
HCT116 and SW480 were transfected with varying amounts of miR-133a mimics and siRNA by reverse transfection according to the manufacturer’s instructions and plated at a density of 4 × 103 cells per well in 96-well plates for cell viability assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO, USA) assay. For each treatment group, triplicate wells were analyzed for cell viability.
Cell invasion assays and wound-healing assay
To measure cell invasion, matrigel-coated invasion chambers (8 µm; BD Biosciences, Franklin Lakes, NJ, USA) were used according to the manufacturer’s instructions. The cells were suspended in a serum-free medium at a density of 5 × 104 cells/mL. A 2-mL of cell suspension was pipetted into the upper chamber of the matrigel-coated inserts. RPMI-1640 medium with 10% fetal bovine serum was added to the lower chamber as a chemoattractant. The cells were incubated for 24 h at 37°C and then, the cells on the upper side of the membrane were removed with a cotton swab. The cells that had invaded the lower surface were fixed with methanol and stained with 0.1% crystal violet. The number of cells that had invaded through the matrigel was counted in five fields for triplicate membranes at ×200 magnification using an inverted microscope (Olympus, Tokyo, Japan).
To evaluate cell motility, a wound-healing assay was carried out. Cells were plated on six-well plates at a density of 2 × 105 cells per well, and wounds were generated using a micropipette tip. Then, cells were rinsed three times with phosphate-buffered saline (PBS) and fresh culture medium was added. The residual gap widths were evaluated from photomicrographs after 96 h of wound establishment. The experiment was performed in triplicate wells for three times.
Colony formation assay
HCT116 and SW480 cells (5 × 104/well) were plated on 24-well plates and transfected with pre-miR-133a, control or co-transfected with miR-133a mimics, and eIF4A1. After 24 h, the cells were collected and seeded (1000–1500/well) on fresh six-well plates for 14 days. Surviving colonies containing at least 50 cells were recorded after fixed with methanol/acetone (1:1) and stained with 5% crystal violet. The experiment was carried out in triplicate wells for three times.
Western blotting and immunohistochemistry
Antibodies against eIF4A1 (ab31217, Abcam, Cambridge, MA, USA) and GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA), and horseradish peroxidase–conjugated secondary antibody (Abcam) were prepared. Tissues of formalin-fixed paraffin-embedded surgical specimens were constructed. Detection of eIF4A1 was performed on paraffin sections (4 µm) with the anti-eIF4A1 antibody (1:200). Then, analyzed by an image analysis software Image-Pro Plus 6.0 (Media Cybernetics Company, Rockville, MD, America).
Western blotting was performed as described previously. Proteins were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). Antibodies against eIF4A1 (1:1000) and GAPDH (1:1000) were probed, and horseradish peroxidase–conjugated secondary antibody was used for further probe. The proteins were visualized using enhanced chemiluminescence (ECL) reagents (Pierce, Rockford, IL, USA). Proteins were quantified using GAPDH as loading control.
Luciferase reporter assay
The fragments, including the 3′-UTR-WT or 3′-UTR-MUT regions of eIF4A1 and a mutant reporter (Luc-eIF4A1-mu), in which the predicted miR-133a binding site on eIF4A1 was mutated, were cloned into luciferase reporters and co-transfected with either a miR-133a mimic or a control. After 24 h of transfection, the firefly and renilla luciferase activities were determined with a dual-luciferase reporter assay system (Promega Corporation, Fitchburg, WI, USA) in accordance with the manufacturer’s suggestions.
In vivo tumorigenicity
Animal experiments were done according to the Institutional Animal Care and Use Committee (IACUC) protocol approved by the Wenzhou Medical University for Use and Care of Animals. HCT116 stable clones with inducible miR-133a and negative control (NC) miRNA were injected (1 × 106) into the left flank of the female BALB/c nude mice (4 weeks old). Mice were segregated into control and experimental groups randomly (N = 6/group) for each animal study. Tumor size was measured every 3 day using caliper, and the tumor volume (V) was calculated as (l × w × w)/2, with l indicating length and w indicating width. Mice were killed by cervical dislocation on day 28, and the tumors were excised and snap frozen for the next test.
Statistical analysis
All data were presented as the mean ± standard error of the mean (SEM), differences between groups were compared using a one-way analysis of variance (ANOVA), with post hoc Student LSD (least significant difference) pairwise comparisons applied as appropriate. For all behavioral test analysis, two-way repeated measures ANOVA with post hoc Bonferroni analysis between groups, where appropriate, was utilized. Significant differences were considered for p < 0.05. Statistical calculations were carried out with Prism software (San Diego, CA, USA).
Results
miR-133a is frequently downregulated in primary CRC and colon cancer cell lines
We first employed quantitative reverse transcription PCR (qRT-PCR) to detect miR-133a levels in 51 CRC and corresponding NTs. The results showed that the average expression level of miR-133a was downregulated in CRC tissues and upregulated in NT tissues (p < 0.001; Figure 1(a)). Further analysis showed that miR-133a expression was negatively associated with the histological grade of colon cancer (Figure 1(b)).

miR-133a was downregulated in the CRC clinical specimens and human CRC cell lines. (a) miR-133a expression was determined by qRT-PCR in 51 pairs of CRC tissue (T) and the corresponding adjacent nontumor tissue (N). (b) Assessment of miR-133a levels from the total RNA derived from CRC tissues according to the tumor stage. (c) The relative expression of miR-133a in the CRC cell lines compared with the human benign colon cells NCM460.
Next, expression of miR-133a in CRC cells (HCT116, HT29, and SW480) was examined. The data showed the miR-133a expression level was downregulated in CRC cell lines compared with a normal NCM460. Taken together, the results demonstrated that lack of miR-133a expression in colon cancer is positively related to the advanced stage, metastasis, and poor prognosis.
miR-133a suppresses colon cancer cell growth in vitro and in vivo
To explore the potential tumor suppressive effect of miR-133a, pre-miR-133a was transfected into colon cancer cell lines (HCT116 and SW480). We performed real-time PCR to check the expression level of miR-133a and pre-miR-133a mimics transfected miR-133a. Ectopic expression of miR-133a in HCT116 and SW480 cells caused a significant decrease in cell viability (p < 0.001 in both cell lines; Figure 2(a) and (b)). Next, the inhibitory effect of miR-133a on cancer cell growth was further confirmed by colony formation assay. miR-133a also induced HCT116 and SW480 cell inhibition by colony formation assay, which could be aborted by the overexpression of eIF4A1 (Figure 3). To further confirm the growth inhibitory effect of miR-133a on colon cancer cells, a xenograft tumor growth assay was performed. The subcutaneous tumor growth curve of HCT116 stably expressed miR-133a or control in vivo was shown in Figure 4. The tumor volume was significantly lower in miR-133a nude mice as compared to the control mice (p < 0.01). These results provided further evidence that miR-133a plays a tumor suppressive role in colon cancer.

miR-133a inhibited colon cancer cell growth in vitro. (a) Ectopic expression of miR-133a in colon cancer cell lines HCT116 and SW480 were evidenced by qRT-PCR after transfection of pre-miR-133a. (b) miR-133a significantly inhibited HCT116 and SW480 cell viability. Cell growth rates were detected by MTT assay.

Effect of miR-133a on HCT116 and SW480 cell functions is mediated by eIF4A1. miR-133a inhibited HCT116 and SW480 cells colony formation, and overexpression of eIF4A1 in cells transfected with pre-miR-133a mimics can counteract the miR-133a effects. Quantitative analysis of HCT116 and SW480 colony numbers (N = 3, mean ± SEM).
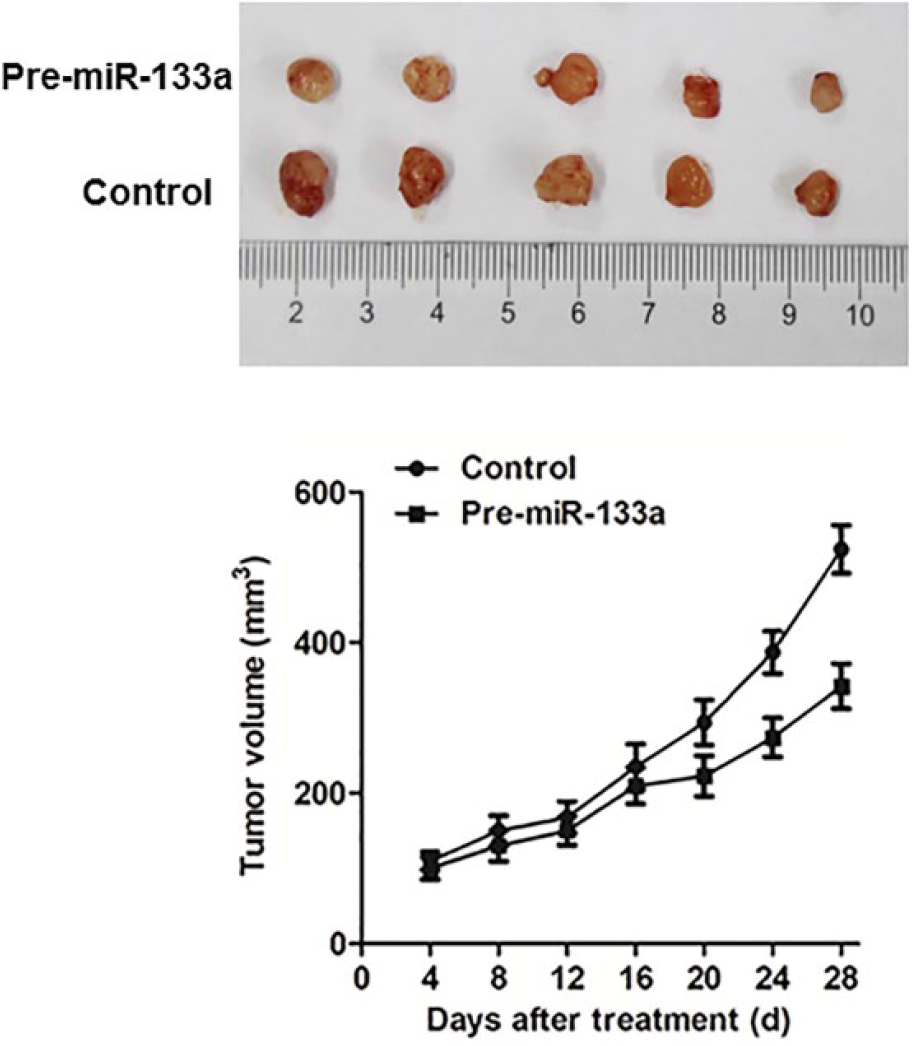
miR-133a inhibited colon cancer cell growth in vivo. Tumor growth curve of pre-miR-133a and control transfected HCT116 cells in nude mice. Data are mean ± SEM (N = 6/group). Images of the tumors induced by pre-miR-133a or control are shown.
miR-133a suppresses CRC cell migration and invasion of colon cancer cell lines
To evaluate the impact of miR-133a on cell invasion and migration, matrigel invasion and wound-healing assays were employed. As shown in Figure 5(a), HCT116 and SW480 cells were transfected with pre-miR-133a or control; induction of miR-133a significantly decreased the invasive cells in HCT116 and in SW480 compared to control cells.

miR-133a inhibited colon cancer cell migration and invasion. (a) Re-expression of miR-133a in HCT116 and SW480 cells significantly inhibited cell invasion ability as determined by cell invasion assay. (b) The inhibition on cell migration by miR-133a was further confirmed by wound-healing assay. The results shown are representative of three independent experiments.
The reduction in wound closure by miR-133a further confirmed its effect in suppressing cell migration. Furthermore, wound-healing assay data showed significantly reduced widths for the residual gaps obtained in the miR-133a transfection and negative control groups. This experiment was repeated for three times.
miR-133a targets eIF4A1 via binding to its 3′UTR
We then investigated the mechanisms by which miR-133a inhibits colon cancer progression. Bioinformatic analysis showed that eIF4A1 was directly suppressed by miR-133a (Figure 6(a)). To test the specific regulation through the predicted binding sites, we constructed a reporter vector which consists of the luciferase coding sequence followed by the 3′UTR of eIF4A1 (Luc-eIF4A1-3′UTR). Wild type (Luc-eIF4A1-3′UTR) or mutated sequence (Luc-eIF4A1-mut 3′UTR) within the putative binding sites was cloned into the pMIR-REPORT vector. Co-transfection experiments in HCT116 cells showed that miR-133a significantly decreased the luciferase activity of Luc-eIF4A1-3′UTR (p < 0.05), but this was not observed in Luc-eIF4A1-mut 3′UTR (Figure 6(b)). Our data thus demonstrated that eIF4A1 was a direct target of miR-133a.

eIF4A1 is a direct target of miR-133a in colon cancer. (a) Human eIF4A1 3′UTR binding site for miR-133a. (b) miR-133a targeted the wild-type but not the mutant 3′UTR of eIF4A1. The data are mean ± SEM. (c) Ectopic expression of miR-133a downregulated eIF4A1 mRNA expression in HCT119 and SW480 cells as determined by qRT-PCR. (d) miR-133a decreased eIF4A1 protein level in HCT116 and SW480 by western blot. (e) Expression of eIF4A1 is significantly upregulated in primary colorectal tumors compared with their adjacent normal tissues (N = 51, left panel). Representative immunohistochemical staining of eIF4A1 in CRC tissue (T) and the corresponding adjacent nontumor tissue (N; right panel).
To further confirm that miR-133a targets eIF4A1, pre-miR-133a or control was transfected into HCT116 and SW480 cells. Transfection of pre-miR-133a resulted in significant reduction of eIF4A1 mRNA and protein expression by qRT-PCR (Figure 6(c)) and by western blot (Figure 6(d)), respectively.
eIF4A1 is upregulated in primary CRC tumors and inversely correlated with miR-133a expression
To assess the importance of eIF4A1 in primary CRC, we compared the level of eIF4A1 expression in 51 paired tumor and adjacent normal tissues. The expression of the eIF4A1 mRNA was significantly increased in CRC tumors compared to the adjacent normal tissues (p < 0.001; Figure 6(e)). eIF4A1 was overexpressed in 86% (44/51) of tumors compared with their normal counterparts.
To further study the relationship between miR-133a and eIF4A1 in human CRC, we measured the protein expressions in 25 paired CRC and adjacent normal tissues using immunohistochemistry. From the results, CRC tissues exhibited a significant eIF4A1 expression compared to the adjacent normal tissues.
Discussion
As one of the most malignant tumors, the median survival time for CRC patients is approximately 22 to 24 months, with a 5-year survival rate of <5%. 10 It is the third most common cause of cancer mortality worldwide and was the fourth most commonly diagnosed cancer in women and men in the United States in 2015. 11 An increasing number of studies have demonstrated that miRNAs may serve as novel diagnostic or prognostic markers or as future therapeutic targets in patients with CRC. 12 In this study, we revealed downregulated expression of miR-133a in CRC tissues compared with matched adjacent NTs. Interestingly, a panel of colon cancer cell lines also showed reduced miR-133a levels compared with a normal colon epithelial cell line. We therefore characterized the putative tumor suppressive function of miR-133a in human colon cancer cell lines. Moreover, additional studies proved that the restoration of miR-133a levels in colon cancer cell lines HCT116 and SW480 significantly inhibited cell proliferation, as evidenced by cell viability and colony formation assays. Recent studies have also shown that miR-133a regulates many different target genes in various cancer cells. For example, fascin actin-bundling protein 1 (FSCN1) was found to be required for miR-133a-mediated changes in breast cancer migration and invasion capacity. 13 Chen et al. 14 found that miR-133a inhibits cellular proliferation, invasion, and metastasis, which are mediated by direct interaction between miR-133a and insulin-like growth factor 1 receptor (IGF-1R) in human osteosarcoma cell lines. Tao et al. 15 found that miR-133a reduced the migration and invasion of prostate cancer cells by directly targeting and inhibiting the expression of epidermal growth factor receptor (EGFR) and its signaling pathway. In nude mice, colon cancer cells overexpressing miR-133a displayed a significantly lower growth rate than that of control cancer cells.
miRNAs have been evaluated most intensively in the field of oncological research, and emerging evidence suggests that altered miRNA regulation is involved in the progression of many tumors, including CRC.16–18 After demonstrating a crucial role for miR-133a in suppressing CRC development, we sought to identify the possible gene effectors involved. Among the miRNA-predicted target genes, we found that eIF4A1 acts as a critical effector of miR-133a. eIF4A1, an adenosine triphosphate (ATP)-dependent RNA helicase, is crucial for assembly of the translational active ribosome. 19 eIF4A1 can unwind RNA secondary structures in the 5′-UTR of mRNAs, which is necessary for efficient binding of the small ribosomal subunit and subsequent scanning of the mRNA sequence for the initiator codon. 20 In addition to cap-dependent translation, eIF4A1 has also been reported to play an important role in cell proliferation and viral DNA replication. 21 The inhibitory effect of miR-133a on cell proliferation can be abolished by overexpression of eIF4A1. Moreover, we have shown that miR-133a significantly represses the luciferase activity of Luc-eIF4A1-3′UTR by targeting the 3′UTR of eIF4A1 mRNA. In CRC patients, both the eIF4A1 mRNA and protein expression levels were significantly increased. These results suggest that miR-133a selectively inhibits eIF4A1 in the progression of CRC.
Recent studies have reported that miR-133a can serve as a potential prognostic marker for esophageal squamous cell carcinoma. 22 The clinical value of miR-133a in CRC remains controversial. It was reported that a higher level of miR-133a is associated with a more aggressive form of CRC, whereas a recent study suggested that a lower level of miR-133a was associated with CRC disease progression and metastasis in a cohort of CRC patients. In our study, miR-133a was not found to be associated with clinicopathological features, including the survival of CRC patients. The clinical value of miR-133a requires further investigation in larger CRC cohorts.
In conclusion, our data provide new evidence supporting the tumor suppressive function of miR-133a in CRC. We also determined that eIF4A1 is a novel target of miR-133a in CRC. In the future, miR-133a may be used as a novel diagnostic biomarker and therapeutic target in the treatment of CRC.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (No. 81571395, 81371748, and 81373075).