
Abstract
The high incidence of esophageal cancer in Northeast India and the unique ethnic background and dietary habits provide a great opportunity to study the molecular genetics behind esophageal squamous cell carcinoma in this part of the region. We hypothesized that in addition to currently known environmental risk factors for esophageal cancer, genetic and epigenetic factors are also involved in esophageal carcinogenesis in Northeast India. Therefore, in this study, we explored the possible association between the two important G1 cell cycle regulatory genes p16 and p53 and environmental risk factors and risk of esophageal carcinogenesis. A total of 100 newly diagnosed esophageal cancer cases along with equal number of age-, sex-, and ethnicity-matched controls were included in this study. Methylation-specific polymerase chain reaction was used to determine the p16 promoter methylation status. Single-nucleotide polymorphism at codon 72 of p53 gene was assessed by the polymerase chain reaction-restriction fragment length polymorphism method. Aberrant methylation of p16 gene was seen in 81% of esophageal cancer cases. Hypermethylation of p16 gene was not found in healthy controls. p53 Pro/Pro genotype was found to be a risk genotype in Northeast India compared with Arg/Pro and Arg/Arg. p53 variant/polymorphism was significantly associated with esophageal cancer risk in the study population under all three genetic models, namely, dominant model (Arg/Pro + Pro/Pro vs Arg/Arg odds ratio = 2.25, confidence interval = 1.19–4.26; p = 0.012), recessive model (Arg/Arg + Arg/Pro vs Pro/Pro odds ratio = 2.35, confidence interval = 1.24–4.44; p = 0.008), and homozygous model (Pro/Pro vs Arg/Arg odds ratio = 3.33, confidence interval = 1.54–7.20; p = 0.002). However, p53 variant/polymorphism was not statistically associated with esophageal cancer risk under the heterozygous model (Pro/Pro vs Arg/Pro). In the case-only analysis based on p16 methylation, the p53 variant/polymorphism (Pro/Pro or Arg/Pro) showed significant association for esophageal cancer risk (odds ratio = 3.33, confidence interval = 1.54–7.20; p = 0.002). Gene–gene and gene–environment interaction using the case-only approach revealed a strong association between p16 methylation, p53 single-nucleotide polymorphism, and environmental factors and esophageal cancer risk. Cases with p16 methylation and p53 variant/polymorphism (Pro/Pro or Arg/Pro) along with both betel quid and tobacco chewing habit (odds ratio = 8.29, confidence interval = 1.14–60.23; p = 0.037) conferred eightfold increased risk toward esophageal cancer development. This study reveals a synergistic interaction between epigenetic, genetic, and environmental factors and risk of esophageal cancer in this high-incidence region of Northeast India. The inactivation of either p16 or p53 in a majority of esophageal cancer cases in this study suggests the possible crosstalk between the important cell cycle genes.
Introduction
Cancer burden is increasing at an alarming rate globally. The global cancer statistics states that there are approximately 456,000 new esophageal cancer cases and 400,000 cancer deaths worldwide during the year 2012. 1 International comparison of cancer incidence in the population-based cancer registries of India revealed that there is a very high incidence of esophageal cancer in the Northeastern part of India.2–4 Meghalaya (East Khasi Hills District) in the Northeast India has the highest incidence of esophageal cancer. 3 The incidence of esophageal cancer in Northeast India is increasing but with a very poor prognosis. Epidemiological study revealed the influence of environmental factors like tobacco, and betel nut chewing and tobacco smoking as the prime risk factors.5,6 However, the molecular pathogenesis for the abnormally high incidence of esophageal cancer in this part of India is not much documented.
Cancer is abnormal growth of undifferentiated cells which might be due to defect in cell cycle regulation. 7 The cell cycle is regulated by the cell cycle checkpoints. The cell cycle genes maintain the genomic integrity of the cells during DNA replication of cell cycle. 8 Deregulation of the cell cycle leads to tumorigenesis, and genetic and epigenetic changes affecting cell cycle genes are the major events during carcinogenesis. 9 The p16 (pRb/p16/cyclin D) and the p53 (p14/MDM2/p53) pathways are the two major cell cycle pathways that are frequently found to be mutated. 10 Defect in these cell cycle pathways leads to uncontrolled proliferation of damaged DNA leading to tumor development.11,12
p16 gene, located at 9p21, prevents cell cycle progression by disrupting the cyclin D/CDK4 kinase complex, thus preventing phosphorylation of retinoblastoma (Rb) protein. 13 Hypophosphorylation of Rb in turn prevents release of transcription factors necessary for progression from G1 to S phase of the cell cycle. 14 The tumor suppressor gene p53, located on the short arm of chromosome 17, plays a pivotal role in cell cycle pathways by controlling cell proliferation. 15
p53 protein induces G1 cell cycle arrest or apoptosis in response to DNA damage. 16 Activation of the p53 network due to cellular stress results in activation of cyclin-dependent kinases which phosphorylate p53 resulting in release of MDM2 with simultaneous stabilization of p53. 17 p53 binds to p21 resulting in its transactivation. p21 then interacts with the cyclin-dependent kinases which prevent Rb phosphorylation and thereby prevent cell proliferation until DNA is repaired. 18
Thus, p16 and p53 are essential G1 regulatory genes of the cell cycle pathway whose loss of function is associated with tumor development. 19 Genetic and epigenetic alterations in this pathway lead to inactivation of these genes thus leading to cancer.20–22
Aberrant DNA methylation in the normally unmethylated promoter region of p16 gene is a critical mechanism for p16 gene inactivation and is commonly associated with silencing of the corresponding gene which results in tumorigenesis including esophageal carcinogenesis. 23 Previous reports have revealed that p16 gene is frequently found to be methylated in a number of cancers.24–26 Similarly, p53 alterations, including p53 mutations and single-nucleotide polymorphism (SNP) in codon 72 of p53 gene, have been reported to be associated with esophageal cancer development.27–29 The SNP at position 72 (R72P) of the p53 protein encodes either an arginine or proline, which can alter the apoptotic activity of p53 via transcriptional and non-transcriptional mechanisms.16,30
Therefore, in this study, we aimed to explore the possible association between the two important G1 cell cycle regulators in this high-incidence region of Northeast India. Moreover, we also hypothesized that there may be a possible gene–gene and gene–environment interaction leading to the abnormally high incidence of esophageal cancer in Northeast India.
Materials and methods
Sample collection and DNA preparation
A total of 100 newly diagnosed esophageal cancer cases along with 100 age (±5 years)-, sex-, and ethnicity-matched controls were included in this study. We collected 3 mL of blood from histopathologically confirmed esophageal cancer cases who visited Aditya Hospital, Dibrugarh, and Assam Medical College and Hospital, Dibrugarh, Assam, India. People from different places of Northeast India like Meghalaya, Manipur, Assam, Arunachal Pradesh, and Mizoram visit these hospitals for diagnosis and treatment. Blood from age (±5 years)-, sex-, and ethnicity-matched healthy controls were also collected from the community. The blood samples were stored at −80°C until further processing. Written informed consent was obtained from all subjects for participation in the study. Socio-demographic characteristics including information on dietary habits, alcohol consumption, and betel nut and tobacco chewing and smoking habit were obtained in a pre-designed questionnaire. The Institutional Ethical Committee of Regional Medical Research Centre, NE Region (ICMR) approved the study design.
DNA extraction
The genomic DNA was extracted from the samples using the Qiagen DNeasy® Blood and Tissue Kit (Qiagen, Hilden, Germany) following the protocol provided.
Bisulfite modification
Bisulfite modification of DNA results in conversion of unmethylated cytosine bases to uracil, whereas methylated cytosines remain unchanged. Upon bisulfite modification, methylated DNA sequence differs from unmethylated DNA, which is used to design methylation-specific primers. 1 µg of genomic DNA was modified using the EpiTect Bisulfite Kit (Qiagen, Hilden, Germany) according to the manufacturer’s specifications.
p16 methylation status by methylation-specific polymerase chain reaction
The methylation status at the promoter region of p16 gene was assessed by methylation-specific polymerase chain reaction (PCR; MSP) as described by Wang et al. 31 The reaction was carried out in a final volume of 25 µL PCR reaction with 100 ng of bisulfite-treated DNA, 1.5 µL of each primer (Sigma Aldrich, St. Louis, MO, USA), 4.5 µL of water, and 12.5 µL DreamTaq Master Mix (Fermentas, MA, USA). The primers used for MSP were methylated primer 5′-TTATTAGAGGGTGGGGCGGATCGC-3′ (sense) and 5′-GACCCCGAACCGCGACCGTAA-3′ (antisense) at an annealing temperature of 65.5°C, and unmethylated primer 5′-TTATTAGAGGGTGGGGTGGATTGT-3′ (sense) and 5′-CAACCCCAAACCACAACCATAA-3′ (antisense) at an annealing temperature of 60°C. EpiTect Methylated Control DNA from Qiagen was used as a positive control, and water was used as a negative control. PCR products were loaded onto 2% gels, stained with ethidium bromide, and directly visualized in gel documentation system (Cell Biosciences, Inc., Santa Clara, CA).
p53 genotyping using PCR-restriction fragment length polymorphism
Genotyping of p53 gene polymorphism was done by PCR amplification using gene-specific primers followed by restriction fragment length polymorphism (RFLP). The PCR amplification for p53 genotype was carried out by a MasterCycler gradient thermo cycler (Bio-Rad, CA, USA) in a final volume of 25 µL containing 150 ng of each primer (Sigma Aldrich, St. Louis, MO, USA), 50 ng genomic DNA, 2 mM MgCl2 (Roche, Basel, Switzerland), 200 µL deoxynucleotide triphosphates (dNTPs; Roche Basel, Switzerland), and 1.5 unit of Taq DNA Polymerase (Roche, Basel, Switzerland). Primer sequences for PCR amplification were 5′-TTGCCGTCCCAAGCAATGGATGA-3′ and 5′-TCTGGGAAGGGACAGAAGATGAC-3′ which produced a 199-bp band. 31 Amplifications for p53 genotype were performed under the following conditions: 96°C for 5 min and 30 cycles of 94°C for 1 min (denaturation), 55°C for 1 min (annealing), and 72°C for 1 min (extension), followed by 72°C for 4 min. After amplification of the PCR product, it was digested at 55°C for 3 h with BstU1 restriction enzyme. The RFLP product was then electrophoresed in 2.5% agarose gel and then visualized in gel documentation system (Cell Biosciences, Inc., Santa Clara, CA). Arg/Arg genotype yields two small fragments of 113 and 86 bp. Pro/Pro genotype yields single 199 bp. The heterozygote Arg/Pro genotype has three fragments of size 199, 113, and 86 bp (Figure 1(a)). Genotyping of 10% of the randomly selected cases and controls was confirmed by sequencing, and 100% concordance was observed (Figure 1(b)).

(a) RFLP photograph of 2% agarose gel electrophoresis representing p53 genotype. Lane M represents 100 bp DNA ladder. Lanes 1, 3, and 4 were characterized by 199, 113, and 86 bp representing Arg/Pro genotype. Lanes 2, 5, and 6 representing 113 and 86 bp represents Arg/Arg genotype. Lane 7 was characterized by single 199 bp representing Pro/Pro genotype. (b) Representative genotypes of p53 codon 72 polymorphisms by sequencing.
Statistical analysis
Multiple logistic regression analysis was used to analyze the data. The conditional maximum likelihood method was used to estimate the parameters of the regression model because of the matched design, and significance was set as p ≤ 0.05 (two-tailed). Difference in distribution of demographic characteristics and methylation and genotype frequencies between cases and controls were evaluated using the chi-square (χ2) test, Fisher’s exact test, and independent samples t-test, wherever appropriate. The crude measure of association between single putative risk factors and esophageal cancer was expressed by deriving odds ratio (OR) and its corresponding 95% confidence interval (CI) calculated from the standard error of the regression coefficient. To control for confounding variables such as betel quid chewing, tobacco chewing, tobacco smoking, alcohol consumption, and other covariates, the data were analyzed by conditional multiple logistic regression to evaluate the extent of risk association.
The strength of the association between p53 codon 72 polymorphism and esophageal cancer risk was measured by ORs with 95% CIs under four genetic models, namely, dominant model (Pro/Pro + Arg/Pro vs Arg/Arg), recessive model (Pro/Pro vs Arg/Arg + Arg/Pro), homozygous model (Pro/Pro vs Arg/Arg), and heterozygous model (Pro/Pro vs Arg/Pro).
ORs with 95% CIs were used to assess the strength of association between the p16 methylation status along with p53 variant/polymorphism and esophageal cancer risk. Further to estimate the risk of esophageal cancer imparted by the synergistic effect of p53 variant/polymorphism and other exogenous factors such as betel quid chewing and tobacco chewing, stratified analysis was performed by making possible combinations among these covariates on the basis of methylation status among esophageal cancer patients. p53 Pro/Pro or Arg/Pro was used in stratified analysis rather than only Pro/Pro in order to increase the power of the laid model.
Tests for Hardy–Weinberg equilibrium (HWE) in both cases and controls were conducted using observed genotype frequencies and a χ2 test featuring one degree of freedom. All statistical analyses were performed using the statistical package SPSS version 17.
Results
Socio-demographic characteristics of the subjects under study
Hundred newly diagnosed esophageal cancer cases (ages ranging from 40 to 75 years; with mean age of 59.98 ± 8.24 years) were enrolled in the study. In all, 66% of the cases were males and 34% were females. The distribution of socio-demographic characteristics of the subjects and risk of esophageal cancer is represented in Table 1.
Socio-demographic characteristics and risk of esophageal cancer.

SD: standard deviation.
Based on Fisher’s exact test.
For independent samples t-test.
Based on chi-square test.
Betel quid and tobacco chewing habit was found to be common among the esophageal cancer cases (91% were betel quid chewers, p < 0.001; 89% were tobacco chewers, p < 0.001). In all, 52% cases were smokers and 50% were alcohol users. The distribution of betel quid and tobacco chewing frequency among the cases was found to be statistically significant (Table 1).
p16 promoter methylation profile and clinicopathological characteristics of esophageal cancer patients
Aberrant promoter methylation of the p16 gene was detected in 81 of 100 (81%) esophageal cancer cases (Figure 2). Agarose gel picture depicting methylation status of cases in representative samples is shown in Figure 3.
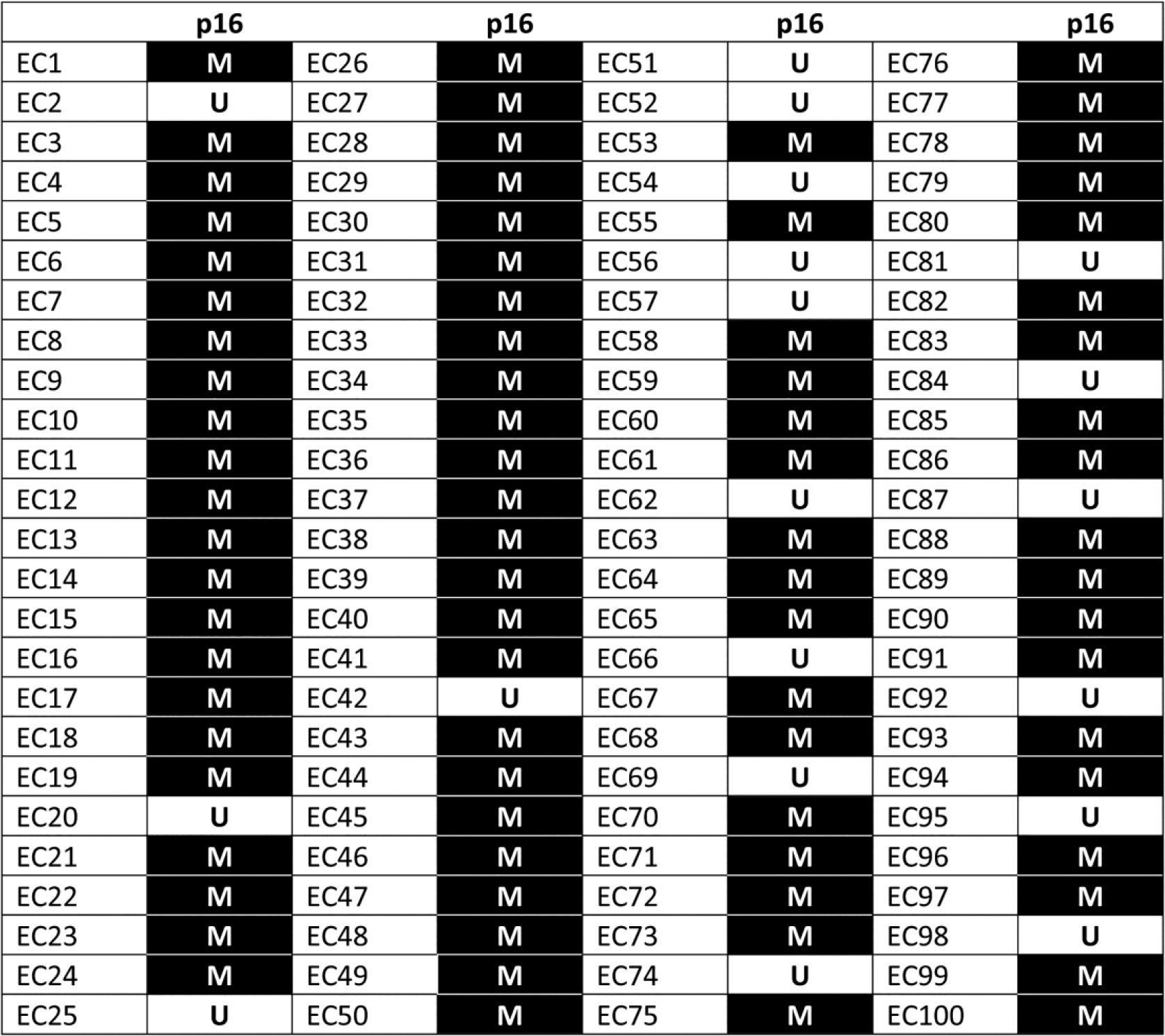
Promoter methylation profile of p16 gene of 100 esophageal squamous cell carcinoma (ESCC) patients. Each column and row represents the gene indicated on top and individual patients. The number indicated on the left corresponds to the patient number. Black rectangles are methylated (M) samples and white rectangles are unmethylated (U) samples.

Photograph of 2% agarose gel electrophoresis showing representative results of methylation-specific PCR analysis for p16 gene. Lanes 1 and 8 are 100 bp ladder. Lanes 9–15 are methylated bands in cases. Lane 16 is Epitect methylated DNA positive control from Qiagen. Lane 17 is negative control (water) for methylated DNA. Lane 4 is positive control for unmethylated DNA from Qiagen. Lane 2 is negative control (water) for unmethylated DNA. Lanes 5 and 6 represent unmethylated bands in controls.
After methylation analysis of all the samples, clinicopathological data were correlated with these results. Age, pathological grade, and tumor topography were not significantly correlated with p16 methylation in esophageal cancer. There was a difference in terms of gender indicating that p16 gene was more frequently methylated in case of males compared with females. This may be justified by the fact that tobacco and betel nut chewing containing nitrosamines which are significant risk factors for esophageal cancer was found to be more among the males than females. Moreover, p16 methylation was seen more in esophageal cancer patients in the age group of 61–70 years. On the contrary, aberrant p16 methylation was found in esophageal cancer patients at all clinical stages, suggesting that these cancers could be methylated at an early stage (Table 2).
p16 promoter methylation profile and clinicopathological characteristics of esophageal cancer patients.

Based on Fisher’s exact test.
Based on chi-square test.
Distributions of p53 genotype and risk of esophageal cancer
Frequency distributions of Arg/Arg, Arg/Pro, and Pro/Pro genotypes among esophageal cancer cases were 20%, 43%, and 37%, while those of controls were 36%, 44%, and 20%, respectively. The association of p53 codon 72 variant/polymorphism and esophageal cancer risk is shown in Table 4. The results reveal that p53 Pro/Pro genotype was found to be significantly associated with increased risk of esophageal cancer among the study population (OR = 3.33, CI = 1.54–7.20; p = 0.002) compared with p53 Arg/Pro and Arg/Arg genotypes. The p53 variant/polymorphism was found to be significantly associated with esophageal cancer under all the three genetic models: dominant model (Arg/Pro + Pro/Pro vs Arg/Arg OR = 2.25, CI = 1.19–4.26; p = 0.012), recessive model (Arg/Arg + Arg/Pro vs Pro/Pro OR = 2.35, CI = 1.24–4.44; p = 0.008), and homozygous model (Pro/Pro vs Arg/Arg OR = 3.33, CI = 1.54–7.20; p = 0.002). However, in cases of the heterozygous model (Pro/Pro vs Arg/Pro OR = 1.89, CI = 0.95–3.76; p = 0.069), p53 variant/polymorphism was not significantly associated with esophageal cancer risk. The distribution of p53 genotype in both cases and controls was in HWE (Table 3).
Distributions of p53 genotype and risk of esophageal cancer.

OR: odds ratio; CI: confidence interval.
Hardy–Weinberg equilibrium test is calculated for 1 degree of freedom and values rounded to two decimals.
Significant.
Interaction of p53 variant/polymorphism with environmental risk factors and risk of esophageal cancer
The association of p53 genotype and environmental risk factors was determined by multivariate logistic regression analysis using both cases and controls. Betel quid and tobacco chewing was found to be significant risk factors for esophageal cancer (p < 0.001). Interactions between p53 genotypes and risk factors were analyzed to look for gene–environment interactions. The interaction of betel quid with p53 genotypes in esophageal cancer showed significant increased risk for p53 variant/polymorphism, namely, Pro/Pro or Arg/Pro and betel quid chewing habit (OR = 5.09, CI = 1.75–14.77; p = 0.003). Similarly, p53 variant/polymorphism, namely, Pro/Pro or Arg/Pro along with tobacco chewing habit conferred a fourfold increased esophageal cancer risk (OR = 4.38, CI = 1.55–12.37; p = 0.005). Gene–environment interaction revealed a cumulative effect of p53 variant/polymorphism (Pro/Pro or Arg/Pro) along with both tobacco and betel quid chewing habit toward esophageal cancer risk in Northeast India (OR = 5.92, CI = 1.89–18.58; p = 0.002) (Table 4).
Interaction of p53 variant/polymorphism with environmental risk factors and risk of esophageal cancer.
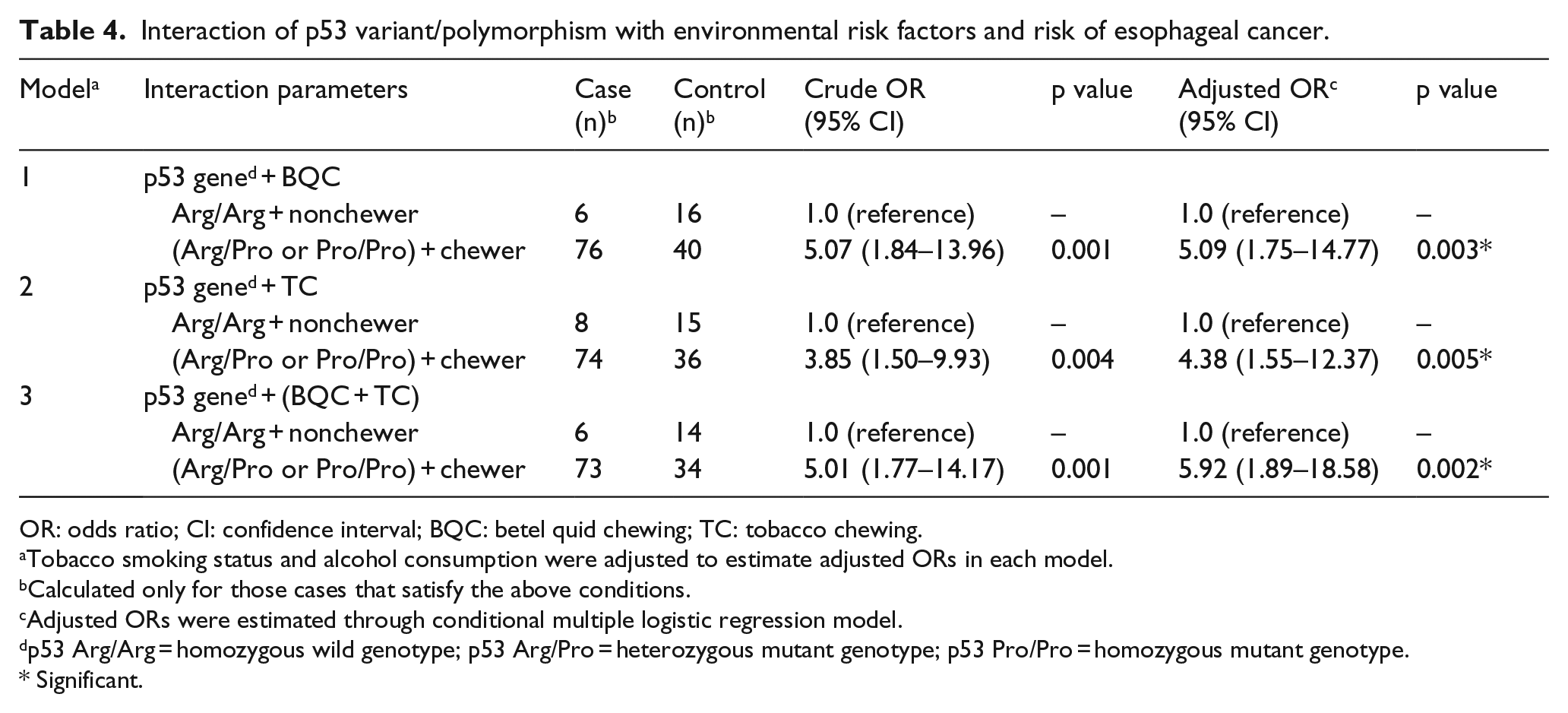
OR: odds ratio; CI: confidence interval; BQC: betel quid chewing; TC: tobacco chewing.
Tobacco smoking status and alcohol consumption were adjusted to estimate adjusted ORs in each model.
Calculated only for those cases that satisfy the above conditions.
Adjusted ORs were estimated through conditional multiple logistic regression model.
p53 Arg/Arg = homozygous wild genotype; p53 Arg/Pro = heterozygous mutant genotype; p53 Pro/Pro = homozygous mutant genotype.
Significant.
Stratified analysis of p53 codon 72 variant/polymorphism and environmental risk factors on the basis of p16 promoter methylation profile of esophageal cancer patients
Since p16 methylation was not seen in healthy controls, a case-only approach for analysis was adopted for gene–gene and gene–environment studies. The esophageal cancer patients included in the study were first stratified based on their p16 methylation profile. In all, 70 out of 81 esophageal cancer patients with p16 promoter methylation and p53 codon 72 variant (either p53 Pro/Pro or p53 Arg/Pro) showed a significant risk for esophageal cancer development (adjusted OR = 5.60, 95% CI = 1.70–18.39; p = 0.005). Further classifying these cases on the basis of tobacco chewing habit, it was seen that those cases with p16 methylation along with p53 codon 72 variant and tobacco quid chewing habit conferred a sixfold increased risk for esophageal cancer (adjusted OR = 6.40, 95% CI = 1.13–36.22; p = 0.036; Table 5).
Stratified analysis of p53 codon 72 variant/polymorphism and environmental risk factors on the basis of p16 promoter methylation profile of esophageal cancer patients.

OR: odds ratio; CI: confidence interval; BQC: betel quid chewing; TC: tobacco chewing.
Tobacco smoking status and alcohol consumption were adjusted to estimate adjusted ORs in each model.
Calculated only for those cases that satisfy the above conditions.
Adjusted ORs were estimated through conditional multiple logistic regression model.
p53 Arg/Arg = homozygous wild genotype; p53 Arg/Pro = heterozygous mutant genotype; p53 Pro/Pro = homozygous mutant genotype.
Significant.
Still, further stratifying these cases, it was observed that cases with p16 methylation and p53 codon 72 variant/polymorphism along with both betel quid and tobacco chewing habit conferred a eightfold increased risk for esophageal cancer development (adjusted OR = 8.29, 95% CI = 1.14–60.23; p = 0.037; Table 5).
Discussion
In this study, we tried to assess the possible interaction and association of epigenetics and genetics with environmental factors and the risk of esophageal cancer in the high-incidence region of Northeast India.
Epigenetics has been defined as the stable alterations in gene expression with no underlying modifications in genetic sequences.32,33 Epigenetic silencing of tumor suppressor genes by aberrant DNA methylation has been found to be a major event in tumorigenesis.34,35 DNA methylation is a process of addition of methyl group at the fifth carbon of the cytosine ring resulting in transcriptional gene inactivation and chromosomal instability.9,36 DNA methylation is found to be an early event in carcinogenesis and can be detected in many kinds of body fluids. 37 Hence, it is of great importance in the field of biomarker discovery. 38
In this study, we have found a high frequency of p16 hypermethylation among esophageal squamous cell carcinoma patients. Our study is in line with earlier reports on p16 methylation and cancer. 25 A slight higher frequency in our study explains the plausible influence of environmental factors like exposure to carcinogenic risk factors in our study population. Northeast Indian population is a mixed population comprising of migrating population from the southeastern Asia. 39 There is a wide disparity in the ethnicity and the dietary habits in this mixed population with exposure to potent carcinogenic compounds like betel nut and tobacco. 26 This prompted us the need to study the genetic and epigenetic changes and their possible association with environmental risk factors and their susceptibility to esophageal cancer.
In addition to p16 hypermethylation, p53 alterations were also detected in the study population. The p53 Pro/Pro genotype was found to confer a significant risk toward esophageal cancer development compared with Arg/Pro and Arg/Arg genotype. Previous studies on p53 codon 72 polymorphism and risk of esophageal cancer have revealed that the Pro/Pro genotype of p53 is a risk genotype among the Asian population. 27 There are also reports of a differential distribution of p53 genotype among different ethnicities. 40 We therefore tried to assess the possible association between p53 variant/polymorphism (Pro/Pro or Arg/Pro) and risk of esophageal cancer.
When the p16 methylation status was compared with the p53 codon 72 polymorphism in esophageal cancer patients, we observed that those cases with p16 methylation and p53 variant/polymorphism (Pro/Pro or Arg/Pro) conferred a significant risk for esophageal cancer.
Although p16 hypermethylation has been reported to be a predisposing epigenetic trait in esophageal squamous cell carcinoma (ESCC), the role of environmental factors cannot be ruled out. Our previous epidemiological study on the identification of possible risk factors for esophageal cancer in Northeast India revealed that high-level exposure to polycyclic aromatic hydrocarbons present in betel nut and tobacco may contribute to the high incidence of esophageal cancer in this region.5,6 Concomitant p16 promoter hypermethylation along with p53 codon 72 polymorphism may be a consequence of environmental, dietary, and lifestyle factors peculiar to this region leading to increased susceptibility to ESCC. We therefore explored the possible association of epigenetic and genetic events along with environmental factors.
For gene–environment interaction, cases with p53 variant/polymorphism (Pro/Pro or Arg/Pro) and tobacco chewing habit were stratified on the basis of p16 methylation. Cases with p16 methylation, p53 variant/polymorphism and tobacco chewing habit conferred a sixfold increased risk for esophageal cancer. Still, further stratifying these cases, p16 methylation and p53 variant/polymorphism along with both betel quid and tobacco chewing habit conferred an eightfold increased risk for esophageal cancer development in Northeast India.
To our knowledge, this is the first report on the possible association of p16 hypermethylation with p53 codon 72 polymorphism and environmental factors in this high-incidence region of Northeast India. These observations may highlight the occurrence of combined molecular mechanisms, which may play a pivotal role in ESCC development.
All these findings in addition to correlation with p16 promoter hypermethylation and p53 codon 72 polymorphism indicate a possible overlap and crosstalk between the involved pathways thus conferring an increased risk of esophageal cancer in Northeast India. Cumulative effect of p16 methylation and p53 variant/polymorphism along with both tobacco and betel quid chewing habit and esophageal cancer risk revealed a strong correlation between epigenetic, genetic, and environmental factors in this high-incidence region of Northeast India. Therefore, p16 gene along with p53 SNP can be used as potential molecular targets for early esophageal cancer detection. Further gene–environment studies of various cancer-related genes in a large sample size would help to elucidate the molecular changes occurring in ESCC and tumor progression in this high-risk population. This would also help in the development of biomarkers for early detection of cancer. Those cases found positive for the above risk factors could be stratified from the population and can be called in for timely endoscopic surveillance and can be routinely monitored for early diagnosis of esophageal cancer. This is of utmost importance to both clinicians and society because cancer can be prevented only if detected early. Therefore, multicentric validation of the study in large samples would give more probable factors for esophageal cancer detection and prevention.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Indian Council of Medical Research (ICMR), Department of Health Research, Government of India under the extramural funding of Northeast Initiative.