
Abstract
Gastric cancer is one of the most common malignancies and leading causes of cancer-related death worldwide. An increasing number of evidence has revealed that gastric tumorigenesis is a multistage pathological state, and epigenetic alterations are considered to play critical roles in the etiology of gastric cancer. Lysine-specific demethylase-1, a histone demethylase, has been linked to malignancy in several human cancers and considered to epigenetically regulate many tumor suppressor genes during tumorigenesis and cancer progression. However, its role and underlying targets in gastric cancer are still unclear. In this study, we detected the lysine-specific demethylase-1 expression level in gastric cancer tissues and cell lines and investigated the function and mechanism of lysine-specific demethylase-1 in the gastric cancer. The in vitro analysis shows that knockdown of lysine-specific demethylase-1 significantly inhibits gastric cancer cell proliferation, migration, and invasion and induces cell cycle G1 phase arrest and cell apoptosis. In vivo assays determine that lysine-specific demethylase-1 downregulation represses gastric cancer cell tumorigenesis. Mechanistic investigation reveals that tumor suppressor KLF2 is a key downstream target of lysine-specific demethylase-1 in gastric cancer. These findings indicate that lysine-specific demethylase-1 is an important oncogene in gastric cancer, and lysine-specific demethylase-1-mediated epigenetic repression of KLF2 plays a critical role in gastric cancer development and progression, which supports lysine-specific demethylase-1 as a potential therapeutic target in this disease.
Introduction
Gastric cancer (GC) is one of the most common cancers and a leading cause of the cancer-related death worldwide. 1 Although lots of efforts have been made to improve the survival of GC patients, the 5-year overall survival (OS) rate is still unsatisfactory that is less than 30%, which is due to the delayed diagnosis, limited treatment options, and recurrence.2,3 In spite of the great advancement on the GC pathogenesis, the regulators and molecular mechanisms that contribute to GC development and progression are still poorly documented. 4 Given the global poor survival rates of this disease, there is an urgent need to identify new factors involved in GC and develop novel diagnostic markers and effective therapies.
Recently, an increasing number of studies have confirmed that GC tumorigenesis is a multistage pathological process that arises from environmental factors, and genetic or epigenetic alterations are considered to play important roles in the development of this disease.4–6 The oncogenic activation of β-catenin and K-ras and amplifications of c-ERBB2 and c-MET genes have been found in GC. 7 Besides, mutations in the tumor suppressor genes are also involved in GC, such as APC (APC, WNT signaling pathway regulator) mutations that are frequently observed in gastric adenomas and only rarely in GCs. 8 In addition to those well-characterized genetic alterations, epigenetic changes, including promoter DNA hypermethylation, and histone modification are also involved in human GC. Hypermethylation of hMLH1 gene promoter is the main mechanism responsible for microsatellite instability (MSI) in GC, while the promoter hypermethylation of p16 is also common in GC. 9 However, the role of histone modification in GC development and progression is not well known.
The histone lysine-specific demethylase-1 (LSD1), also known as KDM1A, is the first identified histone demethylase. 10 Generally, LSD1 serves as a core component of transcriptional co-repressor complex CoREST complexes through mediating the demethylation of H3K4m1/m2 in diverse biological settings.11,12 Mounting studies have revealed that LSD1 plays important roles in diverse fundamental cellular processes including cell growth, differentiation, epithelial–mesenchymal transition, and invasion.13–15 Additionally, LSD1 is frequently overexpressed and has been found to function as a bona fide oncogene in a broad spectrum of human cancers.16,17 For example, aberrant LSD1 overexpression in oral squamous cell carcinoma (OSCC) was associated with tumor aggressiveness and shorter OS, and siRNA-mediated knockdown of LSD1 in OSCC cells resulted in impaired cell proliferation, invasion, and induced cell apoptosis and enhanced chemosensitivity to 5-fluorouracil (5-FU). 18 However, the role of LSD1 in GC is still not well known.
In this study, we evaluated the LSD1 expression in GC tissues and cells and found that LSD1 is overexpressed in GC tissues and cells. Moreover, its functions in GC cell proliferation, cell cycle progression, apoptosis, migration, and invasion were determined by integrating RNAi approaches using GC cell lines and animal models. Mechanistic investigation was performed to document by which mechanism LSD1 regulates its underlying target KLF2 in GC cells. Our findings highlight that LSD1 critically contributes to GC tumorigenesis as well as aggressive phenotype, suggesting the great potential of LSD1 as a novel diagnostic marker and therapeutic target for GC patients.
Materials and methods
GC specimens and cell lines
A total of 20 paired GC tissue samples and the corresponding adjacent noncancerous tissues were obtained from Yishui Hospital between 2011 and 2012 with informed consent. Patients were diagnosed with GC based on histopathological analysis without local or systemic treatment before surgery. This study was approved by the Research Ethics Committee of Yishui Hospital. Four GC cell lines BGC823, SGC7901, MGC803, and AGS and a normal gastric epithelium cell line GES1 were purchased from the Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, China). SGC7901, AGS, and GES1 were cultured in Dulbecco’s modified Eagle’s medium (DMEM); BGC823 and MGC803 cells were cultured in RPMI 1640 with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA).
RNA extraction and quantitative reverse transcription polymerase chain reaction analysis
The total RNA of GC tissue samples and cells was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Then, 1 µg RNA was reverse transcribed into complementary DNA (cDNA) (20 µL) with random primers or oligo dT using the PrimeScript RT reagent Kit (TaKaRa, Dalian, China). For quantitative real-time PCR (qPCR) assays, SYBR Premix Ex Taq (TaKaRa) was used and the assays were carried out on Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The primers used for detecting LSD1 and target genes are shown in Supplementary Table 1. The results of quantitative reverse transcription polymerase chain reaction (qRT-PCR) were analyzed and normalized to the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression level and then converted to fold changes.
siRNA transfection
The siRNAs for LSD1 or negative control were used to knockdown LSD1 expression in GC cells, and siRNA sequence for LSD1 is listed in Supplementary Table 1. The cells were grown in 6-well plates and transfected with siRNA by RNAiMAX (Invitrogen) according to the manufacturer’s instructions. After 48 h of transfection, cells were harvested for further qRT-PCR or western blot assays.
Cell proliferation and colony formation assays
The cell proliferation ability of GC cells after transfection with LSD1 siRNA or negative control (NC) was assessed using Cell Counting Kit-8 (Dojindo, Shanghai, China) and EdU assay kit (Life Technologies Corporation Carlsbad, CA, USA). The GC cells colony formation capability was examined using colony formation assays.
Cell cycle and apoptosis analysis
BGC823 and SGC7901 cells transfected with LSD1 or NC siRNA were harvested 48 h post-transfection. For cell cycle analysis, GC cells were stained with propidium iodide (PI) using the CycleTEST™ PLUS DNA Reagent Kit (BD Biosciences) and analyzed by FACScan. The percentage of cells in G1, S, and G2/M phase were then counted. For cell apoptotic analysis, the GC cells were double stained with PI and FITC-Annexin V using the FITC Annexin V Apoptosis Detection Kit (BD Biosciences). After that, the cells were analyzed with a flow cytometry (FACScan®; BD Biosciences).
Transwell assays
To determine the cell migration and invasive ability, transwell assays were performed in 24-well plate using chambers with 8-µm polycarbonate membrane (Corning Incorporated, Corning, NY, USA). For invasion assays, GC cells transfected with LSD1 siRNA or NC were seeded on the top side of the Matrigel (BD, Franklin Lakes, NJ, USA) pre-coated membrane; for migration assays, cells were seeded on the top side of membrane without Matrigel. The cells on the upper chamber were removed using cottons swabs after incubation for 24 h; then, cells on the lower membrane surface were fixed and stained using 0.5% Crystal violet solution. Cells in six random fields were counted in each well.
Tumor formation assay
To evaluate the role of LSD1 on GC cell tumorigenesis, 3-week-old female athymic BALB/c nude mice were maintained under pathogen-free conditions according to protocols. BGC823 cells transfected with LSD1 shRNA or empty vector were harvested and then re-suspended at a concentration of 1 × 108 cells/mL with phosphate-buffered saline (PBS). Then, a volume of 100 µL suspended cells was injected into each mouse. Once the tumors are formatted, tumor volume was assessed every 3 days. By 18 days after injection, the mice were euthanized, and the volume and weight of each tumor was examined. This study was approved by the Committee on the Ethics of Animal Experiments of the Yishui Hospital.
Western blot assay and antibodies
BGC823 and SGC7901 cells transfected with LSD1 or NC siRNAs were lysed with RIPA extraction reagent (Beyotime, Beijing, China) supplemented with cocktail (Roche, CA, USA). Then, 40 µg total protein was separated by 8% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins on the gel were then transferred to polyvinylidene fluoride (PVDF) membranes (0.22 µm) (Millipore, Billerica, Massachusetts, USA) and incubated with LSD1 (Millipore), KLF2 (Sigma-Aldrich, St. Louis, MO, USA), or GAPDH (Cell Signaling Technology, Danvers, MA, USA) antibodies. Finally, the enhanced chemiluminescence (ECL) chromogenic substrate was used to quantify the protein levels by densitometry (Quantity One software; Bio-Rad, Hercules, California, USA).
Chromatin immunoprecipitation
LSD1 or NC siRNA-tranfected BGC823 and SGC7901 cells were fixed with formaldehyde and to generate DNA–protein cross-links. Then, cell lysates were sonicated to generate chromatin fragments and immunoprecipitated with LSD1 and H3K4me2-specific antibody (Millipore). IgG was used as negative control. Precipitated chromatin DNA was then analyzed by qRT-PCR.
Statistical analysis
The one-way analysis of variance (ANOVA), Mann–Whitney U test, and Student’s t test (two-tailed) were conducted to analyze the in vitro and in vivo data using SPSS 17.0 software. p values less than 0.05 were considered significant.
Results
LSD1 is overexpressed in GC
To examine the LSD1 expression in GC tissues and nontumor tissues, we first performed immunohistochemistry (IHC) staining analysis and found that LSD1 are expressed highly in GC tissues compared with nontumor tissues. Furthermore, an analysis of a cohort of 20 paired GC tumor and adjacent nontumor tissue sample results using quantitative real-time PCR (qRT-qPCR) showed that LSD1 expression levels are more higher in GC tumor tissues (Figure 1(a)). We then assessed the expression level of LSD1 in GC cell lines (SGC7901, BGC823, MGC803, and AGS) and GES1, an immortalized, normal human gastric mucosal cell line, using qRT-PCR. Compared with GES1 cells, BGC823, SGC7901, and MGC803 cells exhibited higher levels of LSD1 expression, while AGS cells showed relative low LSD1 expression level (Figure 1(b)). Next, we knock down LSD1 expression in BGC823 and SGC7901 cells with higher LSD1 expression levels using specific siRNAs. The results of qRT-PCR showed that the LSD1 messenger RNA (mRNA) levels in BGC823 and SGC7901 cells transfection with LSD1 siRNA are significantly decreased compared with control cells (Figure 1(c)). Similarly, western blot assays also showed that LSD1 proteins levels are also remarkably decreased in LSD1 siRNA-treated GC cells (Figure 1(d)).

LSD1 is highly expressed in gastric cancer tissues and cell lines. (a) qRT-PCR analysis of LSD1 expression in 20 pairs of GC tissues and corresponding nontumor gastric tissues. The LSD1 mRNA levels were normalized to GAPDH. (b) qRT-PCR analysis of LSD1 expression in GES1 cell line and four GC cell lines. The LSD1 mRNA levels were normalized to GAPDH. (c) qRT-PCR analysis of LSD1 expression in BGC823 and SGC7901 cells after transfection with LSD1 siRNA or NC siRNA. (d, e) Western blot analysis of LSD1 protein level in BGC823 and SGC7901 cells after transfection with LSD1 siRNA or NC siRNA.
Knockdown of LSD1 expression inhibits GC cell proliferation and colony formation
To evaluate the possible role of LSD1 in GC cell phenotype, we first performed Cell Counting Kit-8 (CCK8) assays in BGC823 and SGC7901 cells after transfected with two different siRNAs for LSD1. The growth curves of BGC823 and SGC7901 cells detected by CCK8 showed that LSD1 knockdown could significantly inhibit GC cell growth (Figure 2(a)). Consistent with these results, the EdU incorporation was also drastically decreased following LSD1 downregulation (Figure 2(b) and (c)). Furthermore, the colony formation assays showed that knockdown of LSD1 significantly inhibited BGC823 and SGC7901 cells colony formation ability. These findings provide evidence that LSD1 has a growth-promoting role in vitro.

Knockdown of LSD1 inhibits cell proliferation and colony formation. (a) Growth curves of BGC823 and SGC7901 cells after transfection with LSD1 or NC siRNA were determined by CCK8 assays. (b, c) BGC823 and SGC7901 cell proliferation ability was evaluated 48 h after transfection with LSD1 or NC siRNA using EdU-incorporation assays. Red, EdU; Blue, nuclear. (d, e) Colony formation assays were used to determine the effect of LSD1 knockdown on the colony formation ability of BGC823 and SGC7901 cells.
Inhibition of LSD1 induces cell cycle progression arrest and cell apoptosis
To further explore the potential mechanisms involving in growth suppression after LSD1 knockdown, we assessed the effect of LSD1 knockdown on cell cycle and apoptosis in BGC823 and SGC7901 cells. The results of cell cycle distribution analysis showed that knockdown of LSD1 significantly increased the percentage of G0/G1-phase cells, indicating that LSD1 downregulation induced a G0/G1 to S-phase arrest in BGC823 and SGC7901 cells (Figure 3(a) and (b)). Furthermore, Annexin V and PI staining revealed that the percentage of apoptotic cells following LSD1 siRNA treatment was drastically increased compared to control cells (Figure 3(b)). These results imply that cell cycle G0/G1 phase arrest and increased apoptosis might contribute to LSD1 knockdown-mediated growth inhibition in GC cells.
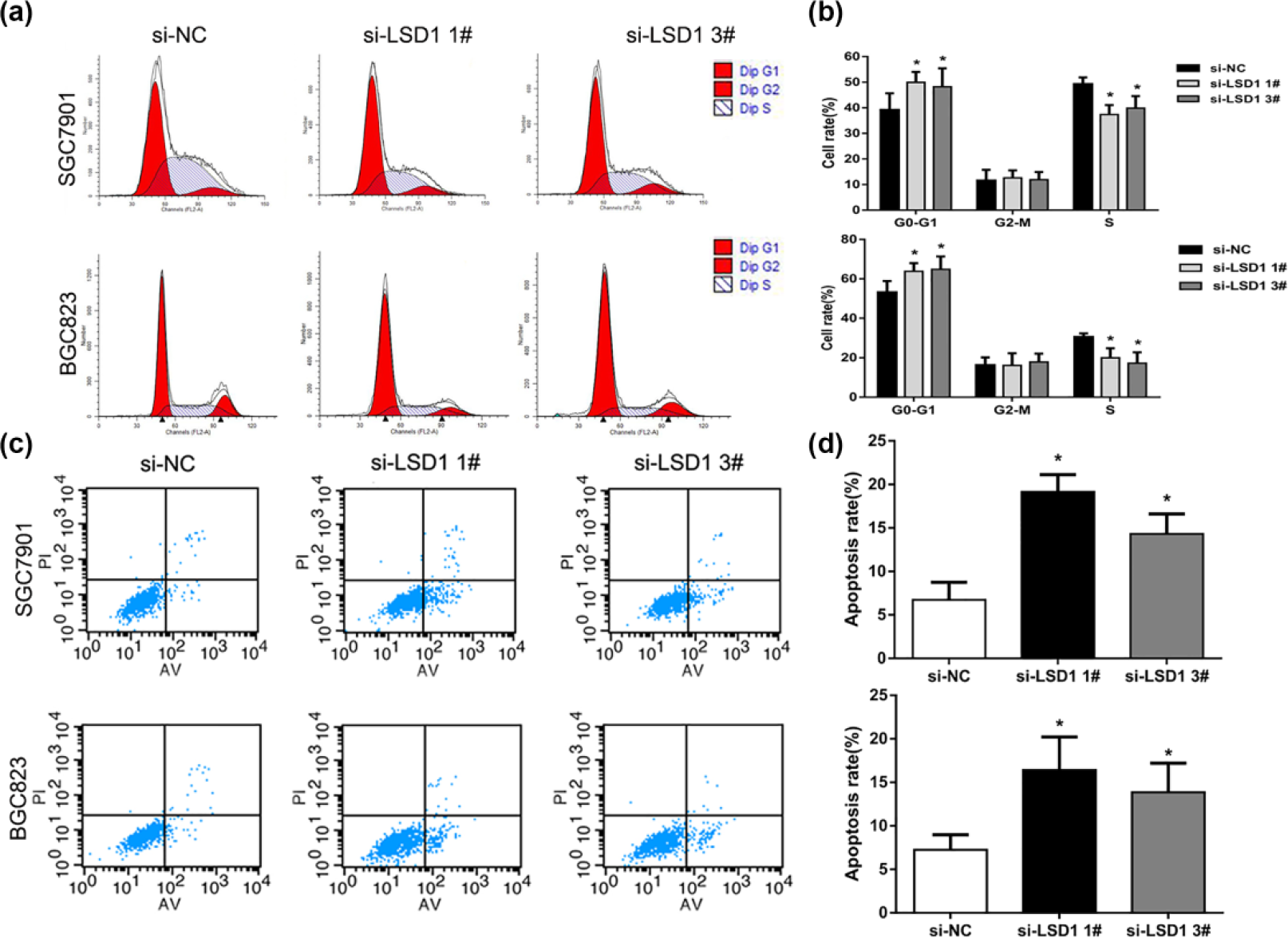
Downregulation of LSD1 induces cell cycle G0/G1 arrest and apoptosis. (a, b) The cell cycle distribution of BGC823 and SGC7901 cells after knockdown of LSD1 was determined by PI staining and flow cytometer analysis. (c, d) The effect of LSD1 knockdown on apoptosis in BGC823 and SGC7901 cells was determined by measuring the percentage of PI and Annexin V-stained cells using flow cytometry.
Decreased LSD1 impaired cell migration and invasion
To further determine whether LSD1 is associated with GC progression, we analyzed the effect of LSD1 knockdown on the migration and invasive behavior of BGC823 and SGC7901 cells. Using the transwell assays, we found that the migration ability of BGC823 and SGC7901 cells was significantly decreased following siRNA-mediated downregulation of LSD1 (Figure 4(a) and (b)). Consistently, the invasive ability of BGC823 and SGC7901 cells transfection with LSD1 siRNAs was also drastically reduced (Figure 4(c) and (d)). Taken together, these results indicate that LSD1 plays important roles in GC development and progression.

LSD1 knockdown impaired cell migration and invasion in vitro. (a, b) The effect of LSD1 downregulation on the migration ability of BGC823 and SGC7901 was analyzed using transwell assays. Data are presented as mean ± SD. (c, d) The effect of LSD1 downregulation on the invasive ability of BGC823 and SGC7901 was analyzed using transwell assays.
LSD1 knockdown inhibits GC cells tumorigenesis in vivo
To further verify the findings of in vitro study, we constructed BGC823 cell lines stably expressing LSD1 siRNA or NC using pGPU6/GFP/Neo vector (Figure 5(a)). Then, the BGC823 cells stably downexpressed or not for LSD1 were subcutaneously injected into nude mice. It is obvious that the tumors formed in LSD1-silencing group grew much slower than those formed in control group (Figure 5(b)). Besides, the tumors weight in the LSD1 knockdown group were reduced significantly compared with that in control group (Figure 5(c)). More importantly, qRT-PCR analysis showed that LSD1 expression levels were significantly decreased in tumor tissues from LSD1 knockdown group compared with that in control group (Figure 5(d)). Moreover, an analysis of IHC revealed that the tumors formed from LSD1 knockdown cells displayed lower Ki-67 signal than those formed from the control cells (Figure 5(e)). These in vivo findings complement the above in vitro data of LSD confirm LSD1 is essential for regulating GC cell proliferation and tumorigenesis.
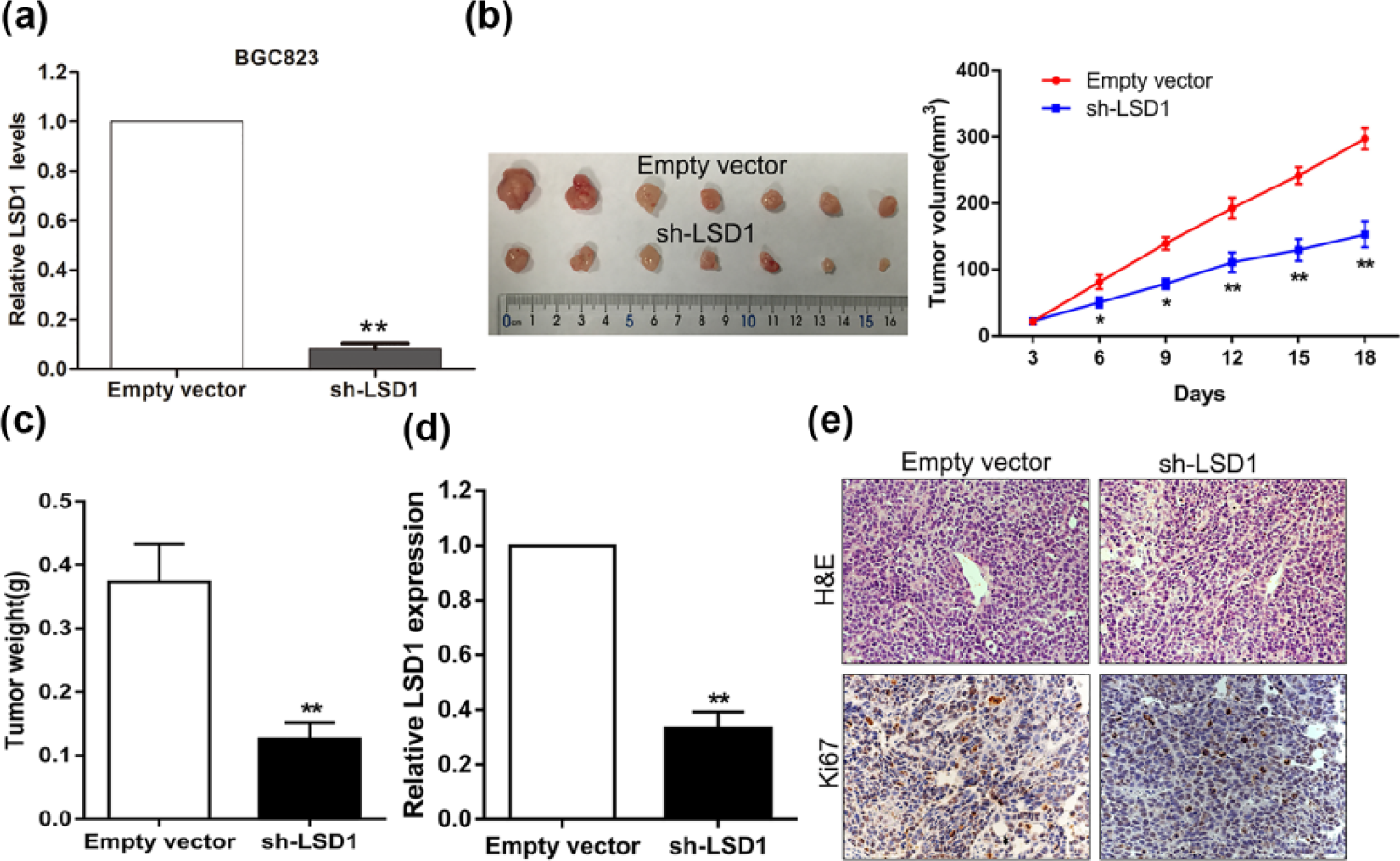
Effect of LSD1 knockdown on tumorigenesis in vivo. (a) BGC823 cells stably expressing LSD1 or negative control siRNA were generated using pGPU6/GFP/Neo. (b) Growth curves of tumors after the injection of BGC823 cells stably knockdown of LSD1 or NC. The volume of tumors was measured every 3 days after inoculation. (c) Tumor weights from LSD1 knockdown and NC group are represented. (d) qRT-PCR analysis of LSD1 expression in tumor tissues formed from LSD1 knockdown and NC group. (e) Tumors developed from LSD1 knockdown BGC823 cells showed lower Ki67 protein levels than tumors developed from control cells.
KLF2 is a key downstream target of LSD1 in GC cells
To explore the potential targets of LSD1 in GC cells, we chose some important tumor suppressors whose expression could be affected by epigenetic alterations to determine which one could be LSD1 target. The results of qRT-PCR analysis showed that LSD1 knockdown could increased the expression of LATS1 and KLF2, but not other genes expression (Figure 6(a)). Similarly, western blot analysis revealed that KLF2 protein levels were increased in LSD1 knockdown BGC823 and SGC7901 cells compared with control cells (Figure 6(b)). To determine whether LSD1 could directly bind to KLF2 promoter region and repress its transcription in GC cells, we designed four pairs of primers across KLF2 promoter region. The results of ChIP assays showed that LSD1 could bind to the KLF2 promoter region and induce H3K4me2 demethylation (Figure 6(c)). Moreover, knockdown of LSD1 reduced its binding to KLF2 promoter regions (Figure 6(d)). These data suggest that KLF2 is a key underlying target of LSD1 in GC cell.

KLF2 is the downstream target of LSD1. (a) qRT-PCR analysis of KLF2, LATS1, and LATS2 expression in BGC823 and SGC7901 cells after transfection with LSD1 siRNA or NC siRNA. (b) Western blot analysis of KLF2 protein levels in BGC823 and SGC7901 cells after transfection with LSD1 siRNA or NC siRNA. (c) ChIP-qPCR of LSD1 occupancy and H3K4me2 binding in the KLF2 promoter in BGC823 and SGC7901 cells, and IgG as a negative control. (d) ChIP-qPCR of LSD1 occupancy and H3K4me2 binding in the KLF2 promoter in BGC823 and SGC7901 cells after knockdown of LSD1, and IgG as a negative control.
LSD1 oncogenic function is partly dependent on repression of KLF2
To confirm whether KLF2 is involved in LSD1 knockdown-mediated suppression of GC cell proliferation and invasion, we upregulated KLF2 expression in SGC7901 cells through transfecting with KLF2 expression vector. The result of western blot showed that KLF2 vector transfection significantly increased KLF2 expression compared with control cells (Figure 7(a)). Next, CCK8 assays indicated that KLF2 overexpression inhibited SGC7901 cell proliferation (Figure 7(b)), and transwell assays showed that increased KLF2 expression impaired SGC7901 cells invasive ability (Figure 7(c)). Furthermore, we performed rescue experiment by co-transfecting with LSD1 and KLF2 siRNAs in SGC7901 cells, and the CCK8 and transwell analysis showed that knockdown of KLF2 could partly reverse LSD1 knockdown-mediated suppression of cell proliferation and invasion (Figure 7(d) and (e)). These data suggest that LSD1 oncogenic function is partly dependent on silencing KLF2 expression in GC cells.

LSD1 promotes GC cells proliferation and invasion partly dependent on repression of KLF2 expression. (a) Western blot analysis of KLF2 expression in SGC7901 cells after transfection with KLF2 or empty vector. (b) Growth curves of SGC7901 cells after transfection with KLF2 or empty vector were determined by CCK8 assays. (c) The effect of KLF2 overexpression on the invasive ability of SGC7901 was analyzed using transwell assays. (d) Growth curves of SGC7901 cells after co-transfection with LSD1 and KLF2 siRNAs were determined by CCK8 assays. (e) The invasive ability of SGC7901 cells after co-transfection with LSD1 and KLF2 siRNAs was analyzed using transwell assays.
Discussion
In the past decades, the majority of traditional molecular studies on GC have focused on identifying novel oncogenes, tumor suppressors, and genetic mutations causing cancer. However, increasing evidence suggests that epigenetic alterations play critical roles in cancer development and progression, including GC.19–21 It has become apparent that the DNA methylation and histone modification are the two major parts of epigenetic alteration. Generally, the DNA methylation or hypermethylation mediated by DNA methyltransferase (DNMT) in the CpG island results in gene silencing; histone modification of lysine methylation at H3K9, H3K27, and H4K20 leads to gene silencing, whereas methylation at H3K4, H3K36, and H3K79 is associated with gene activation. 22 Importantly, recent studies have revealed that several histone modification regulators play a key role in human cancers development and progression. For example, the histone methyltransferase EZH2, a catalytic subunit of PRC2, is highly expressed in numerous cancers including GC. Given its role in tumorigenesis, cancer progression, and prognostic value in several cancer types, EZH2 appears a relevant therapeutic target.23,24
In this study, we identified that histone demethyltransferase LSD1 is overexpressed in GC tissues and cells. Further loss of function assays indicate that knockdown of LSD1 in GC cells could inhibit cell proliferation and tumorigenesis, migration, and invasion ability and induce cell G0/G1 phase arrest and cell apoptosis. These findings suggest that LSD1 might play an important role in GC carcinogenesis and progression. Consistently, several studies also reveal that LSD1 expression is upregulated in other cancer types including non–small cell lung cancer (NSCLC), 15 esophageal cancer, 25 tongue cancer, 26 and colon cancer. 27 Moreover, Shi et al. 28 reported that LSD1 could be recruited by long noncoding RNA LINC00673 and repress NCALD expression through inducing H3K4 demethylation in NSCLC cells, which resulted in promoting NSCLC cells proliferation and tumorigenesis.
Herein, we found that tumor suppressor KLF2 is an important underlying target of LSD1 in GC cells. KLF2 mRNA and protein levels are both significantly upregulated in GC cells after knockdown LSD1. Furthermore, ChIP experiments confirmed that LSD1 directly binds to KLF2 promoter region and mediates H3K4 demethylation modification. KLF2 is one of the prominent members of Kruppel-like factor (KLF) family, which contains Cys2/His2 zinc-finger motifs and functions as the transcriptional repressors or activators to regulate gene expression. 29 Recently, the members of KLF family are considered as the potential oncogenes or tumor suppressors in different cancer types for their effects on cell growth and migration. 30 Numerous studies have demonstrated that KLF2 expression is diminished in multiple cancers, such as NSCLC, 31 and pancreatic ductal adenocarcinoma. 32 Thereby, KLF2 is considered as new tumor suppressor for its inhibitory effect on cancer cell proliferation, metastasis, and promotion of cell apoptosis. Interestingly, the EZH2-mediated H3K27 trimethylation has been found to contribute to the KLF2 expression silence, while our findings reveal that LSD1-mediated H3K4 demethylation also involves in repression of KLF2 transcription.
In summary, our study revealed that the histone demethylase LSD1 is overexpressed in human GC tissues and cells. Knockdown of LSD1 expression exhibits tumor-suppressive effect through inhibition of cell proliferation, migration, invasion, and tumorigenesis and induction of cell cycle G0/G1 arrest and cell apoptosis in GC. LSD1 exerts oncogenic promotion function partly through suppression of tumor suppressor KLF2 expression by mediating H3K4 demethylation. These findings suppose that LSD1 is an important oncogene during human GC development and progression, and its inhibitors may be useful therapy drug for cancer patients.
Footnotes
Acknowledgements
R.F. and F.T. contributed equally to this work and should be regarded as joint first authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.