
Abstract
MicroRNAs are small noncoding RNAs which regulate gene expressions at post-transcriptional level by binding to the 3′-untranslated region of target messenger RNAs. Growing evidences highlight their pivotal roles in various biological processes of human cancers. Among them, miR-138, generating from two primary transcripts, pri-miR-138-1 and pri-miR-138-2, expresses aberrantly in different cancers and is extensively studied in cancer network. Importantly, studies have shown that miR-138 acts as a tumor suppressor by targeting many target genes, which are related to proliferation, apoptosis, invasion, and migration. Additionally, some researches also discover that miR-138 can sensitize tumors to chemotherapies. In this review, we summarize the expression of miR-138 on regulatory mechanisms and tumor biological processes, which will establish molecular basis on the usage of miR-138 in clinical applications in the future.
Introduction
MicroRNAs (miRNAs) are a class of approximately 22 nucleotide-long, small, and noncoding RNA molecules, which can regulate the expression of many important target genes post-transcriptionally through binding to the 3′-untranslated region (3′-UTR) of target messenger RNAs (mRNAs).1–5 Interestingly, some miRNAs also have been found to regulate gene expression by targeting coding region directly. 6 As we all know, a miRNA can regulate multiple mRNAs, and similarly, each mRNA contains potential target sites for numerous miRNAs. More and more evidences suggest that miRNAs are involved in a wide range of biological processes, including cell proliferation, apoptosis, metastasis, invasion, and even chemoresistance.7–10 Among the abundant miRNAs, miR-138 has recently emerged as a considerable tumor suppressor in multiple malignancies, such as cervical cancer, osteosarcoma, non-small-cell lung cancer (NSCLC), ovarian cancer (OC), and gallbladder carcinoma.11–14 Specifically, miR-138 inhibits proliferation, induces apoptosis, restrains both metastasis and invasion, and enhances chemosensitivity of tumor cells through the suppression of multiple targets.7,15–18 All these indicate that miR-138 can display various functions by targeting sorts of genes in biological processes of different cancers.
Here, we will focus on regulatory mechanisms of miR-138 expression and the network of genes which are associated with the functions of miR-138 in the cancer progression and treatment. Consequently, given the expression pattern and functions, miR-138 can bring a novel guidance in molecular targeting treatment of cancers in future.
The regulation of miR-138 expression
MiR-138 is highly conserved among vertebrates. 19 Two primary transcripts, pri-miR-138-1 and pri-miR-138-2, are encoded on chromosome 3 (3p21) and 16 (16q13), respectively, and generate the mature miR-138.20,21 Deletion of these chromosomal locations are frequently detected in thyroid carcinomas, and loss of heterozygosity at both of them is confirmed in oral squamous cell carcinoma (OSCC), which discloses that biological functions in these cancers may be associated with downregulation of miR-138.22–24 Currently, the mechanisms on miR-138 dysregulation in human cancers have been reported, and evidences suggest that epigenetic mechanism, transcriptional factor regulation, hormonal regulation, and other molecules are involved25–28 (Figure 1).
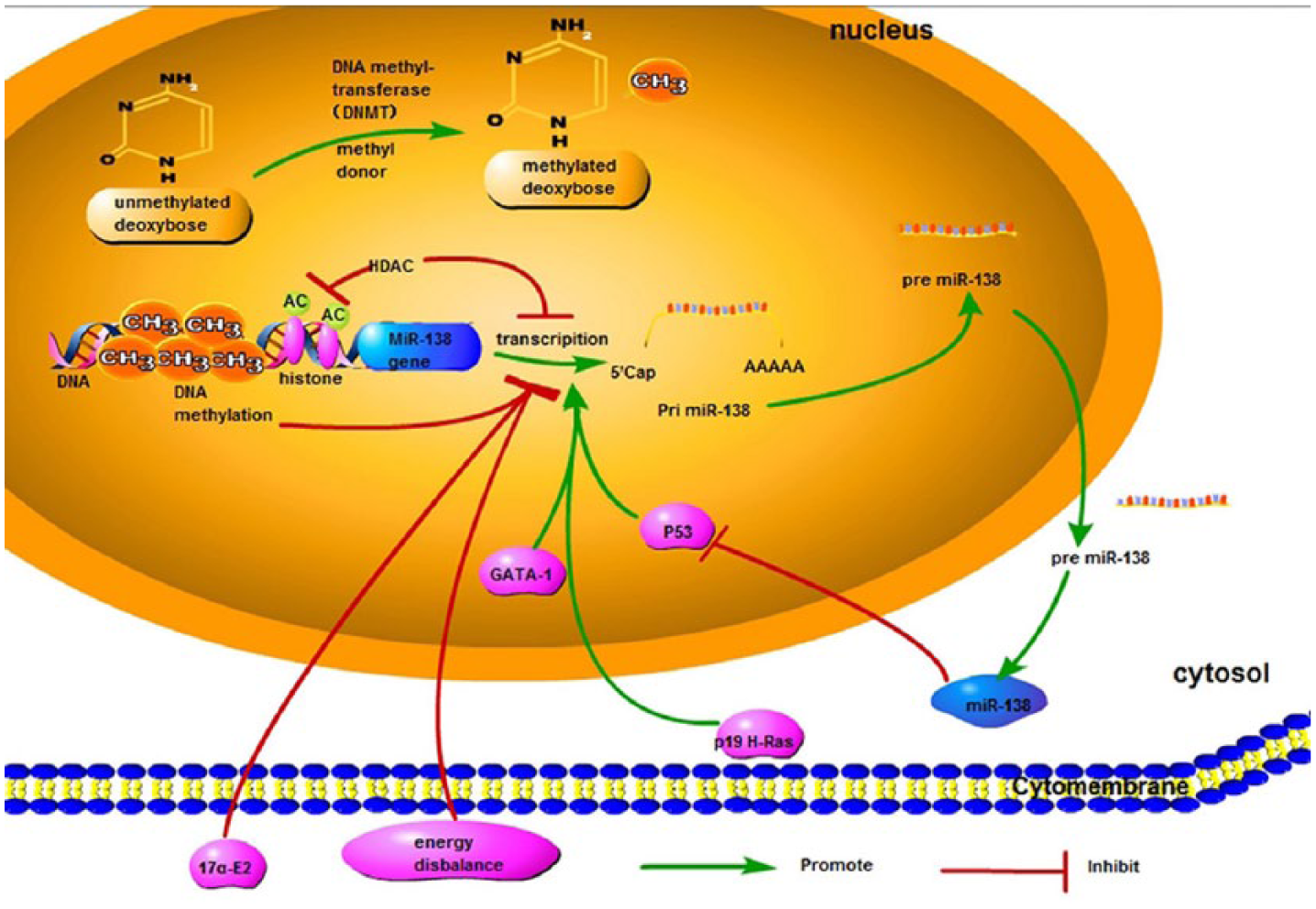
Summarization of how miR-138 expression is regulated. Transcriptional factors such as P53 and GATA1 drive the expression level. P19 H-Ras, a member of Ras oncogenes family, has also been shown to increase miR-138 expression. In contrary, epigenetic mechanisms including DNA methylation and deacetylation of histones decrease the expression of miR-138. Hormonal regulation shows that miR-138 is downregulated by 17a-E2, and energy disbalance is also involved in regulating the expression level of miR-138.
DNA methylation, one of epigenetic mechanisms, has been demonstrated to regulate miRNA expression.29,30 Many tumor suppressor miRNAs are silenced by aberrant hypermethylation such as miR-203, miR-155, miR-137, miR-193a, and miR-200c.29–32 As for miR-138, miR-138-2 shows higher methylation variation in human breast samples compared to miR-138-1, and it is further identified to have a negative correlation between its expression and DNA methylation.25,31 Similarly, deacetylation of histones has also been considered to be strongly related with expression of miRNAs. Deacetylation of histones has the ability to promote tumorigenesis by repressing genes which can inhibit cancer progression or by inducing expression of genes that advance tumor angiogenesis, migration, and invasion. 33 MiR-138 is downregulated in human head and neck squamous cell carcinoma (HNSCC) cells; however, it is significantly upregulated in suberoylanilide hydroxamic acid (SAHA)-treated HNSCC cells. SAHA is a typical histone deacetylase (HDAC) inhibitor, which can alter expression of genes and affect crucial signaling pathways promoting tumorigenesis. 34 It indicates that miR-138 can be reactivated by HDAC inhibitor.
The expression of miRNAs also has been regulated by P53, one of the various transcription factors (TFs),35–37 and there is a “TF-miRNA feedback loop” in the mutual regulation of p53-miRNAs. 38 A recent study has shown an evident increase of miR-138 after p53 activated in human NSCLC cells, but not in rat and mouse cells. More interestingly, in turn, it is found that p53 can be regulated by miR-138 in mouse and rat cells, but this did not happen in human NSCLC cells. 26 So, the mutual regulation between p53 and miR-138 is confirmed as species specific. Similar to p53, the GATA1 is another TF that drives the transcript expression of miR-138. Over-expressed GATA1 upregulates the expression of pri-miR-138-2 and mature miR-138 in vivo. Furthermore, the expression and transcriptional activity of GATA1 is modulated by BCR-ABL. Moreover, imatinib can inhibit BCR-ABL tyrosine kinase activity and activates GATA1, leading to upregulation of miR-138 transcription. High-level miR-138, in turn, represses expression of both BCR-ABL and hence improves the clinical efficacy of imatinib. 21 Therefore, this regulatory circuit loop exhibits an effect on improving therapeutic response to imatinib.
P19 H-Ras has also been shown to increase miR-138 expression. 27 H-Ras together with N-Ras and K-Ras are three members of the Ras oncogenes family, whose mutation has been implicated in about 30% of all tumors.39–41 P19 H-Ras can induce a G1-/S-phase delay and regulates cell growth and metastasis. 42 Garcia-Cruz et al. 27 found that miR-138 was upregulated by p19 overexpression, suggesting that p19 may have had a sufficient impact on miR-138 expression.
In addition, Hu et al. 28 demonstrated that hormonal regulation contributes to the expression of miR-138 in steroidogenic cells of the testis, ovary and adrenal glands. Concretely, miR-138 is downexpressed in response to 17α-ethinylestradiol (17α-E2) treatment. Notably, 17α-E2 is a synthetic estrogen which can enhance MCF-7 breast cancer cell proliferation. 43 These indicate that hormonal regulation of 17α-E2 on miR-138 expression may be implicated in breast cancer.
Last but not least, the effect of energy balance interventions on miR-138 expression is worth being mentioned. Colon cancer is consistently correlated with obesity 44 and miR-138 is validated significantly downregulated among the diet-induced obesity (DIO) mice, 45 which make us assume that miR-138 may influence colon or other organ carcinogenesis due to energy disbalance.
MiR-138 affects tumor biology
MiR-138 inhibits proliferation
The miR-138 expression is downregulated in various cancers such as anaplastic thyroid carcinoma (ATC), NSCLC, gallbladder carcinoma, and OSCC, indicating that miR-138 may serve as a tumor suppressor in these cancers.14,46–48 More studies have confirmed that the overexpression of miR-138 can significantly inhibit cell proliferation.15,48,49 In detail, miR-138 decreases cell proliferation in NSCLC and osteosarcoma cells by reducing zeste homolog 2 (EZH2) expression level.11,47 Moreover, miR-138 represses tumor proliferation of glioblastoma multiforme through inhibiting EZH2-mediated cyclin-dependent kinase 4/6 (CDK4/6)-retinoblastoma protein (Rb)-E2F1 pathway. 50 E2F1, as a TF, controls the expression of genes involved in cell proliferation and is able to drive EZH2 transcription, making the pathway a signal loop. 51 It is also showed that miR-138 decreases the abilities of proliferation by inhibiting Forkhead box C1 (FOXC1), 52 transcription of which can be repressed by EZH2 in breast cancer cells. 53 Wnt pathway participates in the process of cell proliferation as well. MiR-138 inhibits Wnt target genes, including TCF7, MSI1, and PAX5, which are crucial factors for the regulation of cell proliferation. 54 In addition, lots of other targets which are mediated directly by miR-138 and associated with cellular proliferation have been found in varied tumors.7,12,55,56 Concretely, G-protein-coupled receptor kinase–interacting protein 1 (GIT1) plays a central role in cell growth as well as migration/invasion,57,58 and downregulation of GIT1 by miR-138 inhibits the proliferation of NSCLC cells. 7 3-phosphoinositide-dependent protein kinase-1 (PDK1) and cyclin D3 (CCND3), two other target genes involved in NSCLC cells, inhibit cell proliferation as well.15,59 CCND3, as an important regulatory factor on G1/S phase of cell cycle, which is downregulated by miR-138, also inhibits cell proliferation of osteosarcoma cell line MG-63. 55 In osteosarcoma, differentiated embryonic chondrocyte 2 (DEC2) gene is also verified as a direct target of miR-138 and plays an important role in cell proliferation. 12 In OSCC cells, miR-138 suppresses the proliferation by targeting Yes-associated protein 1 (YAP1) and G protein alpha inhibiting activity polypeptide 2 (GNAI2).48,56 The miR-138 overexpressing OSCC cells exhibits a low growth rate with a decreased expression of YAP1. 48 It is also acknowledged that GNAI2 is a proto-oncogene involved in the initiation and progression of several tumors,56,60 and miR-138 reduces the expression of GNAI2, which results in reduced proliferation. 56 MiR-138 inhibits the expression of Ras homolog gene family, member C (RhoC), Bcl-2-associated athanogene-1 (Bag-1), and c-Met, all of which decrease the abilities of proliferation in different types of tumors cells14,16,49 (Table 1; Figure 2).
The target gene of miR-138 and involvement in biology process in different cancers.

NSCLC: non-small-cell lung cancer; OSCC: oral squamous cell carcinoma; TSCC: tongue squamous cell carcinoma; CC: cholangiocarcinoma; CLL: chronic lymphocytic leukemia; ccRCC: clear cell renal cell carcinoma; BC: bladder cancer; LC: larynx carcinoma; HNSCC: head and neck squamous cell carcinoma; OC: ovarian cancer; CRC: colorectal cancer.

MiR-138 plays an important role in the progression and development of malignant tumors by mediating multiple signaling pathways and various target genes. MiR-138 represses tumor proliferation through EZH2-CDK4/6-pRb-E2F1 signal loop and Wnt pathway including TCF7, MSI1, and PAX5. MIR-138 also regulates the proliferation of cancer cells by inhibiting PDK1, CCND3, YAP1, c-Met, FOXC1, DEC2, and GNAI2. Moreover, the transcription of FOXC1 gene can be repressed by EZH2. MiR-138 induces apoptosis of tumor cells by regulating CD95-mediated apoptosis and intrinsic apoptosis pathway including caspase-3, Bax, and Bcl-2. Also, the GNAI2 is associated with cell apoptosis by regulating the level of Bcl-2. All these demonstrate that miR-138 inhibits cell proliferation and induces apoptosis in various cancers.
MiR-138 induces apoptosis
Cell apoptosis is indispensable in cellular processes and miR-138 promotes cancer apoptosis by regulating several relevant effectors. Resistance toward CD95-mediated apoptosis is an important character of chronic lymphocytic leukemia (CLL) cells. 71 MiR-138 downregulates depalmitoylating enzymes, acyl protein thioesterases 1 and 2 (APT1 and APT2), in CLL, both of which directly interact with CD95 and impair palmitoylation-dependent CD95-mediated apoptosis. 62 Moreover, miR-138 activates extrinsic apoptosis pathway with active caspase-8 and intrinsic apoptosis pathway including caspase-3, Bax, and Bcl-2. 10 In detail, miR-138 increases expression of Bax and decreases expression of Bcl-2, resulting in an increased Bax:Bcl-2 ratio, which activates caspase-3 and finally leads to apoptosis. Besides, Caspase-8 may also be relevant to the alterations in expression of Bax and Bcl-2 so that provides a link between extrinsic and intrinsic pathways of apoptosis.10,72 In osteosarcoma, the cell apoptosis rate is significantly enhanced through downregulating the expression of DEC2 with overexpressed miR-138. 12 Inhibition of the expression of hypoxia-inducible factor-1alpha (HIF-1a), one of the key regulators in cancer cells, can increase apoptosis in clear cell renal cell carcinoma (ccRCC) cells. 17 Interestingly, DEC2 can facilitate HIF-1α stabilization and promotes HIF-1 activation in osteosarcoma, resulting in the progression of human osteosarcoma. 73 Also, upregulated miR-138 can promote apoptosis of tongue squamous cell carcinoma (TSCC) and NSCLC cells by targeting GNAI2 and EZH2, respectively.47,56 It is further thought that the GNAI2 affects the cell apoptosis by regulating the expression of Bcl-2. 74 All these highlight a pivotal role for miR-138 in the regulation of cell apoptosis by targeting its target genes (Table 1; Figure 2).
MiR-138 inhibits invasion and metastasis
Cell migration and invasion are initial steps in tumor cell metastasis. MiR-138 shows a novel role in decreasing the metastatic and invasive activity in sorts of cancers.7,12,13,63,65,75 The epithelial-to-mesenchymal transition (EMT) process, the transforming growth factor-beta (TGF-β) signaling, the RhoC-Erk-matrix metalloproteinase-2/9 (MMP-2/9) pathway, and the cofilin signaling all participate in the tumor cell metastasis16,55,63,69 (Table 1; Figure 3).

As for invasion and metastasis of cancer cells, miR-138 suppresses them through prohibiting the EMT process and affecting many important signaling pathways, including the TGF-β signaling pathway, RhoC-ROCK2-FAK-Src-Erk1/2, RhoC-ERK-MMP-2/9, and Limk1/cofilin signaling pathway. SEMA4C, Vimentin, GIT1, NGAL, TWIST2, and E-cadherin, which is downstream of EZH2, ZEB2, Slug, and SOX4, are all involved in EMT process. MiR-138 inhibits the invasion and migration of cancer cells by regulating their expression. Smad4 is an intermediate in the TGF-β signal pathway, and both Slug and ZEB2 are downstream genes of it, which indicate the close connection between EMT and TGF-β signal pathway. Additionally, some other metastasis-associated genes such as RMND5A, NFκB p65, and DEC2 are all regulated by miR-138 and show inhibitive effect on cancer cells invasion and migration. All the above demonstrate that miR-138 inhibits cell migration and invasion in various cancers.
The EMT is a critical biological process, during which epithelial cells alter their morphology and motile behavior and adopt the ability of increased migration. Therefore, reversal of EMT is an ideal strategy against cancer metastasis.68,76,77 By targeting vimentin and EZH2, overexpression of miR-138 can inhibit EMT process, thereby reducing the invasion of breast cancer cells and HNSCC cells.63,64,68 Moreover, it is identified that EZH2 reduces cell migration and invasion through silencing effects on the downstream genes, E-cadherin (E-cad). 68 Descended osteosarcoma cell migration and invasion are also mainly attributed to miR-138/EZH2. 11 Similarly, miR-138 overexpression inhibits bladder cancer (BC) cell, larynx carcinoma (LC) cell, and HNSCC cell invasion and metastases by targeting ZEB2, which is well-defined as essential EMT mediators and regulates the transcription activity of the E-cad gene.65,66,68 Also, in SKOV-I6, SRY-related high-mobility group box 4 (SOX4) and HIF-1α are demonstrated to be two direct targets functioning as an inhibitor of cell migration and invasion. Furthermore, the downstream mediators of SOX4 and HIF-1α are epithelial growth factor receptor (EGFR) and Slug, respectively. 8 Moreover, SOX4 and Slug are transcriptional repressors of E-cad.78,79 In colorectal cancer (CRC) cells, miR-138 displays the inhibitory effect on the cell migratory ability by targeting TWIST2, an oncogene being valuable as a prognostic marker for CRC and involved in EMT as well.70,80 Overexpression of miR-138 extremely results in decreased cell migration and invasion abilities in NSCLC cell as well, and GIT1 and SEMA4C are identified as two novel targets of miR-138 in the progress of NSCLC EMT. 7 Besides, neutrophil gelatinase–associated lipocalin (NGAL) suppression by miR-138 leads to the loss of the ability to undergo EMT, and cell migration capability is inhibited in endometrial and pancreatic carcinoma cells, AsPC-1, and RL95-2. 81 All these show the critical role of EMT in tumor cell metastasis, and miR-138 has the ability to inhibit EMT so that cancer metastasis is suppressed.
TGF-β signaling can be pro-tumorigenic. Smad4, as one of the intermediates in the TGF-β signal pathway, promotes the migration ability of tumor cells. 82 In MG-63 cells, the expression levels of the target genes, Smad4, and NF-κB p65 are downregulated by miR-138, inducing the suppression of cell invasive ability. 55 Notably, Smad4 can induce transcription of downstream genes including Slug and ZEB2. 83 These results point out that TGF-beta-signaling may be strongly associated with EMT induction and maintenance.
In miR-138 overexpressed HNSCC cell lines, focal adhesion kinase (FAK) activation is considerably depleted by targeting RhoC. Additionally, overexpressing miR-138 causes a reduction of the phosphorylation levels of Src and Erk1/2 and consequently attenuates cell invasion. 67 Moreover, miR-138 can directly target FAK, with significantly less cell invasion in both 293 epithelial kidney cells and HeLa cervical carcinoma cells. 84 RhoC, a member belonging to the Ras homolog (Rho) family GTPases, also plays a significant role in CC cell metastasis involving the inhibition of activation of extracellular signal-regulated kinase (ERK) and the downexpression of MMP-2/9. 16 Except for RhoC, Rho-associated kinases 2 (ROCK2), known as RhoC downstream effector, is another key gene in the Rho GTPase signaling pathway, and overexpression miR-138 also suppresses cell migration and invasion of TSCC by targeting ROCK2.61,85 It is also demonstrated that hepatocellular carcinoma (HCC) invasion and EMT are related with activation of a RhoC-ROCK2-FAK-signaling pathway.86,87
The cofilin signaling is a classic pathway mediating cell metastasis. The upregulation of miR-138 leads to downregulation of its target LIM kinase 1 (Limk1) in OC cells, whose migration and invasion capacities are repressed via the Limk1/cofilin signaling pathway. 69 Besides, RMND5A (required for meiotic nuclear division 5 homolog A) is thought to be related to cell migration too. Overexpression of miR-138 downregulates target RMND5A and reduced Exportin-5 stability, which in turn restored miR-138 expression levels. This feedback loop in the pathway of miR-138, RMND5A, and Exportin-5 may contribute to HeLa cell migration. 88 DEC2 not only enhances cell proliferation but also contributes to the progression and metastasis; therefore, by inhibiting the expression of DEC2, miR-138 exhibits its inhibitory effect on osteosarcoma cell invasion. 12
MiR-138 affects chemotherapy
Chemotherapeutic insensitivity remains a major obstacle. Although the specific regulatory mechanism remains unclear, increasing evidences have suggested that miR-138 is involved in several processes, like drug-induced apoptosis and EMT, which may be closely related to chemosensitivity.11,18,89
As known, apoptosis is a common pathway mediating the killing functions of anticancer drugs. MiR-138 can enhance chemotherapy drug-induced apoptosis so that acting as a tumor suppressor. In MG-63 and U2OS cells, elevated expression of miR-138 significantly upregulates the activity of caspase-3, a key executor of cell apoptosis by targeting EZH2, and enhances osteosarcoma cell chemosensitivity to cisplatin. 11 Besides, Wang et al. 90 found that miR-138 was correlated with DNA damage which affected cellular sensitivity to DNA-damaging agents. MiR-138 directly targets histone H2AX, one crucial player in the repair of DNA lesions, and induces genomic instability after DNA damage. Consequently, miR-138 sensitizes U2OS cells to DNA-damaging agents. 18 In K562 cells, the percentage of apoptotic cells is obviously increased by miR-138 in the presence of imatinib. In contrast, the imatinib-induced apoptosis is attenuated by miR-138 inhibitor. 21 Moreover, downregulated expression of excision repair cross-complementation group 1 (ERCC1) by the enforced increase in miR-138 levels increases sensitivity of A549/DDP cells to cisplatin, and increased apoptosis is assessed. 91 All the results exhibit the importance of miR-138 on drug-induced apoptosis involved in chemosensitivity.
EMT plays a role not only in cancer cell metastasis and invasion but also in cancer cell resistance to chemotherapeutics.89,92 In A549/ADM and NCI-H23/ADM cells, ectopic expression of miR-138 upregulates E-cad and downregulates vimentin, partly through targeting ZEB2, both of which are EMT-related markers, displaying that miR-138 sensitizes NSCLC cells to ADM via reversing EMT process. 89 Furthermore, adenosine triphosphate (ATP)-binding cassette subfamily B member 1 (ABCB1) has a relationship with cancer stem cell–like (CSC) properties and EMT and mesenchymal-to-epithelial transition (MET) inhibitors, 92 which have been administered to NSCLC patients in clinical trials.93,94 Inhibition of ABCB1 by miR-138 overcomes acquired resistance to MET inhibitors in NSCLC cell lines. 92 All these make miR-138 a novel therapeutic strategy for NSCLC patients with acquired resistance through suppressing EMT.
Many other researches also support that miR-138 increases chemosensitivity; however, the exact mechanisms responsible for the regulatory function are not described detailedly.34,84,95 Han et al. showed that miR-138 upregulation increased cisplatin sensitivity in H460 and SPC-A1 cells through the interaction with downstream target CCND3. 59 In HeLa cells, stable expression of miR-138 sensitizes the cells to 5-fluorouracil (5-FU), doxorubicin, and FAK inhibitor Y15 by downregulation of FAK. 84 In PC9GR and H1975 cells, miR-138 upregulates cellular gefitinib response via downregulation of G protein-coupled receptor 124 (GPR124), so miR-138 can serve as potential therapeutic approach for overcoming NSCLC gefitinib resistance 95 (Table 2).
MiR-138 influences chemotherapies through targeting associated genes.

CML: chronic myeloid leukemia; NSCLC: non-small-cell lung cancer.
Conclusion and perspectives
This review is aimed to summarize the pleiotropic roles of miR-138 in the progression of cancers. Many factors contribute to the regulatory mechanisms of miR-138 expression, including epigenetic mechanism, transcriptional factor regulation, and hormonal regulation. In the above-mentioned diseases, such as NSCLC, OSCC, TSCC, CC, CLL, ccRCC, and HNSCC, miR-138 levels are downregulated, which discloses that it may be considered as a tumor suppressor. Besides, the complex network of genes involved in diverse biological processes, which consist of proliferation, apoptosis, invasion and metastasis, is further highlighted. Additionally, miR-138 is able to sensitize tumors cells to chemotherapies in some tumors. All these suggest that miR-138 serves as a promising biomarker in cancer development and progression.
However, since the same target gene may be regulated by several miRNAs in different cancers and the genes are multiplex even in the same cancer, miR-138 is just only a small part of the complex regulatory network. Moreover, considering the species-specific differences of the regulations of miR-138, the cross-species differences of the regulations of the target genes are still poorly understood. Finally, the specific regulatory mechanism and signaling pathways of miR-138, its targets, and functions are not fully elucidated in many tumors. Therefore, more efforts are needed before the experimental findings of miR-138 can be applied into clinic for treating kinds of cancers.
Footnotes
Acknowledgements
H.-h.S. and D.-d.W. contributed equally to the writing of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was funded by grants from the National Natural Science Foundation of China (81372396).
Statement of human rights
For this type of study, formal consent is not required.
Statement on the welfare of animals
This article does not contain any studies with human participants or animals performed by any of the authors.