
Abstract
Multiple myeloma, a typical hematological malignancy, is characterized by malignant proliferation of plasma cells. This study was to identify differently expressed long non-coding RNAs to predict the survival of patients with multiple myeloma efficiently. Gene expressing profiles of diagnosed patients with multiple myeloma, GSE24080 (559 samples) and GSE57317 (55 samples), were downloaded from Gene Expression Omnibus database. After processing, survival-related long non-coding RNAs were identified by Cox regression analysis. The prognosis of multiple myeloma patients with differently expressed long non-coding RNAs was predicted by Kaplan–Meier analysis. Meanwhile, stratified analysis was performed based on the concentrations of serum beta 2-microglobulin (S-beta 2m), albumin, and lactate dehydrogenase of multiple myeloma patients. Gene set enrichment analysis was performed to further explore the functions of identified long non-coding RNAs. A total of 176 long non-coding RNAs significantly related to the survival of multiple myeloma patients (p < 0.05) were identified. In dataset GSE24080 and GSE57317, there were 558 and 55 patients being clustered into two groups with significant differences, respectively. Stratified analysis indicated that prediction of the prognoses with these long non-coding RNAs was independent from other clinical phenotype of multiple myeloma. Gene set enrichment analysis–identified pathways of cell cycle, focal adhesion, and G2-M checkpoint were associated with these long non-coding RNAs. A total of 176 long non-coding RNAs, especially RP1-286D6.1, AC008875.2, MTMR9L, AC069360.2, and AL512791.1, were potential biomarkers to evaluate the prognosis of multiple myeloma patients. These long non-coding RNAs participated indispensably in many pathways associated to the development of multiple myeloma; however, the molecular mechanisms need to be further studied.
Keywords
Introduction
Multiple myeloma (MM), a typical hematological malignancy accounting for approximately 1% of neoplastic diseases and 13% of hematologic cancers, 1 is characterized by malignant proliferation of plasma cells in the bone marrow microenvironment and monoclonal protein in the blood or urine, accompanied by organ dysfunctions including anemia, hypercalcemia, renal insufficiency, and osteolytic bone disease. 2 It is reported that the annual age-adjusted incidence is 5.6 cases per 100,000 persons in Western countries. The median age at diagnosis is approximately 70 years; herein, 37% of patients are younger than 65 years. Commonly, the survival time (St) of MM patients varies from weeks to 10 years; recently, with the introduction of autologous stem-cell transplantation and the availability of agents such as thalidomide and bortezomib, the overall survival has extended. As for patients younger than 50 years, the relative 5- and 10-year survival has increased to 56.7% and 41.3% in 2002–2004, respectively.3,4 Even though, the prognosis of MM patients is always poor due to the inefficient early diagnosis. Efficacious approaches identifying highly risked MM patients could provide the possibility of timely individual therapy and furthermore improve the prognostic situation and extend the survival time.
Long non-coding RNAs (lncRNAs) with a length of over 200 nt, together with small ncRNAs, belong to non-coding RNAs (ncRNAs) which do not synthesize protein synthesis or encode short polypeptide only. It has been revealed that at least 90% of the human genome is transcribed actively to RNA, but less than 2% of RNA encodes proteins.5,6 Initially, the ncRNAs accounting for 98% of the whole genome were considered as “junk RNA” without biological functions. 7 However, further researches have made it clear that ncRNAs are involved in cell proliferation, metabolism, and movement, as well as the autophagy, apoptosis, and many other physiological processes of cells. 8 Moreover, ncRNAs were identified indispensable for fine tuning the genetic network, and the deregulation of ncRNA was tightly related to the pathogenesis of an increasing number of diseases, especially cancers. 8 As an abundant class of ncRNAs, lncRNA encodes within the genome, and their transcription is regulated dynamically with cell type specificity. 9 Many lncRNAs were found 5′ capped, poly-adenylated, and spliced, like messenger RNAs (mRNAs),10,11 forming a myriad of functional secondary structures,12,13 which participate in important signal-transduction processes such as chromatin modification, transcriptional activation, and the regulation of post-transcription and protein functions. 14 Some lncRNAs also serve as precursors for shorter regulatory ncRNAs (e.g. snoRNAs and miRNAs). 15
Having been highly conserved throughout mammalian evolution including humans, the lncRNAs have been shown to be aberrantly expressed in cancer tissue and to be involved in oncogenic or tumor-suppressive processes. 13 In this way, lncRNA can as well provide a lot of essential information for prediction of survival of multiple types of tumors.16,17 This novel approaches using the data of expression profile have been applied to determine the prognosis of the patients with tumors like breast cancer, 18 colorectal cancer, 19 prostate cancer, 20 and non-Hodgkin’s lymphoma. 21 However, clinical practice of the approach identifies many problems, including over fitting, lack of validation, heterogeneity among patients and tumors, and neglecting current clinical variables. Enlarging the data scale of expression profiles and clinical information might help solve some problems.
In the present research, large scale of expression profiles and clinical statistics of MM were integrated and analyzed to identify the associated lncRNAs. Then, these differently expressed lncRNAs were applied to predict the prognosis of MM patients. The practical approach focusing on the recent progress of understanding the relationship between lncRNAs and prognosis of MM will pioneer the prognostic evaluation of MM.
Materials and methods
Data source and patient information
Gene expression profilings of highly purified bone marrow plasma cells performed in newly diagnosed patients with MM, GSE24080, 22 were downloaded from Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). A total of 558 samples, including 340 cases enrolled on total therapy 2 (TT2) in training set and 214 patients enrolled in total therapy 3 (TT3) in the validation set, were available on the platform of Affymetrix Human Genome (HG) U133 Plus 2.0 Array (GPL570 (HG U133 Plus 2.0)). On the same platform, the gene expression profiles of patients with MM (N = 55) who have been treated previously (GSE57317) were obtained as well.
Data processing
The probe-level data were converted into the corresponding genetic symbols, based on the relationship of the genes and the matching probes on platform. The microarray data in CEL files were converted into expression measures using the random effects meta-analysis (RMA) function of the Affy package in R, 23 and then, standardized microarray data were further processed with Z-score. 24
LncRNA annotation
Genomic and Transcriptomic Explorer (GATExplorer), a database and web platform integrating a gene loci browser with nucleotide level mappings of oligo probes from expression microarrays, 25 was applied to visualize probes in Affymetrix HG U133 Plus 2.0 together with any lncRNAs. With a series of files and packages (R chip definition files (CDFs)) provided by this platform to analyze particular query expression datasets, certain annotation information was available using affy package in Bioconductor (http://www.bioconductor.org/packages/release/bioc/html/affy.html). CDF files of ncRNA mapping to the probes from expression microarrays were downloaded from GATExplorer, from which expression profile data of lncRNA were obtained using ncrnamapperhgu133plus2cdf_3.0. Means of probes matching with multiple lncRNAs were compared to facilitate the further identification and analysis. Cox regression analysis of the relationship between lncRNA expression and patients’ survival time was performed to identify relating lncRNAs. 26 LncRNAs with p < 0.05 were selected to predict the survival time of MM patients. A k-means clustering 27 algorithm of the expression value of selected lncRNAs was applied to divide MM patients into groups, on which Kaplan–Meier analysis was carried out.
Kaplan–Meier survival analysis and stratified analysis
Kaplan–Meier survival analysis 28 was used to estimate the differences of patients’ survival between groups in k-means clustering. Log-rank test 29 was applied to determine whether there are statistical differences between survival curves. All the analyses were based on the R Language 3.2.3 and Bioconductor.
To assure the practicability of the 176 lncRNAs in predicting patient survival, Kaplan–Meier survival analysis of MM patients in two databases (GSE24080 and GSE57317) was carried out separately. In order to evaluate the specificity of the 176 lncRNAs in predicting patient survival independent of other pathological features, stratified analysis 30 was performed based on the concentrations of serum beta 2-microglobulin (S-beta 2m (Sβ2M)), albumin (ALB), and lactate dehydrogenase (LDH) in serum of MM patients. The Cox-proportional hazard model was applied to estimate the association between lncRNA expression and survival times of MM patients.
Gene set enrichment analysis
To further explore the functions of identified lncRNAs in survival predicting, a new version of the Java-based software (GSEA-P 2.0; 31 http://www.broadinstitute.org/gsea) was used to determine concordant differences between two biological states. This new functionality makes it possible for us to directly import gene sets from Molecular Signature Database (MSigDB) for analysis using gene set enrichment analysis (GSEA). Differently expressed lncRNAs between the two groups of 558 patients were selected for GSEA. False discovery rate (FDR) < 0.05 and p values accounting for 1000 hypotheses testing less than 0.05 were the threshold for significant changes. All results of GSEA were visualized by using Cytoscape and Enrichment Map.
Results
Identification of survival-related lncRNAs
A total of 2096 lncRNAs were annotated. Cox-proportional hazard model for single-agent substitution was used to estimate the association between lncRNA expression and survival times of 559 patients with MM in database GSE24080. A total of 176 lncRNAs with p < 0.05 and |Z-score| > 1.8 were identified as significantly related, including 89 with positive correlation and 87 with negative correlation (Figure 1). Herein, top 20 lncRNAs most significantly reflecting the survival situations of MM patients were listed (Table 1).
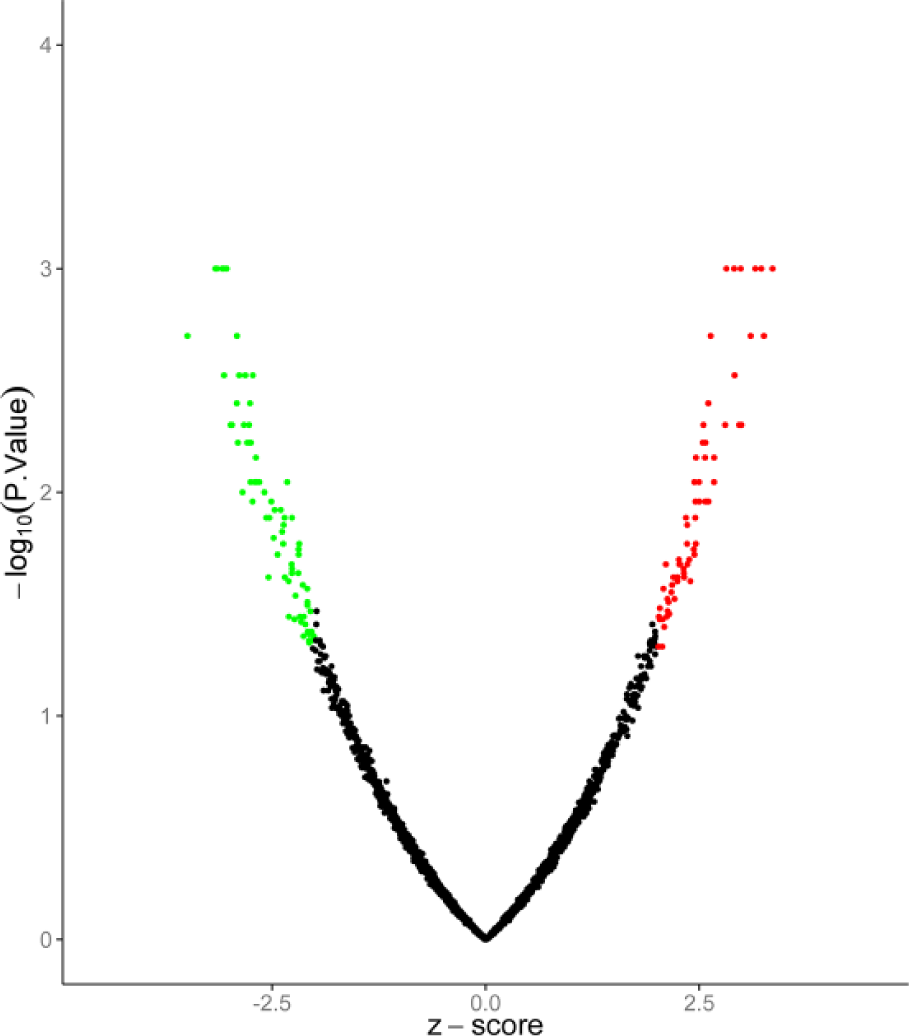
LncRNAs identified as significantly related to survival of MM patients. The green dots represent lncRNAs whose expression level is negatively correlated with the patients’ survival. The red dots represent lncRNAs whose expression is positively correlated, and the black ones represent lncRNAs without correlation. Thresholds are p < 0.05 and |Z-score| > 1.8.
LncRNAs significantly relate to survival conditions of MM patients (top 20).

Kaplan–Meier survival analysis of GSE24080
In all, 558 patients in database GSE24080 were clustered into two groups in k-means clustering analysis based on the expression of 176 lncRNAs (Figure 2(a)). In Kaplan–Meier survival analysis, the overall survival rates of the two groups were significantly different (Figure 2(b)). The p value in log-rank test was 0.0002. The average survival time of patients with good outcomes (87.43 months) was evidently longer than patients with poor outcomes (64.56 months). Similar tendency was shown in disease-free patients with p < 0.0001 in log-rank test (Figure 2(c)).

Kaplan–Meier survival analysis of MM patients in GSE20480: (a) k-means clustering analysis based on the expression of 176 lncRNAs, (b) Kaplan–Meier survival curves of overall survival rates (k = 2), and (c) Kaplan–Meier survival curves of disease-free patients of MM (k = 2).
Survival prediction of patients in independent dataset
To further confirm the effectiveness of identified lncRNAs in predicting the survival of MM patients, the expression of 176 lncRNAs was used to predict survival time of MM patients in another independent dataset GSE57317 (Figure 3(b)). A total of 55 patients with MM available for k-means clustering were divided into two groups with 23 and 32 patients, respectively. Similarly, significant differences in survival rate between the two groups were identified in Kaplan–Meier survival analysis (Figure 3(a)). The average survival time of cluster 1 (17.53 months) was far below that of cluster 2 (27.14 months) in log-rank test (p = 0.0222).

Kaplan–Meier survival analysis of MM patients in GSE57317: (a) k-means clustering analysis based on the expression of 176 lncRNAs and (b) Kaplan–Meier survival curves of overall survival rates (k = 2).
Specificity of the identified lncRNAs
According to the concentration of Sβ2M, 558 patients in GSE20480 were grouped as high risk and low risk. In low-risk group (n = 318, Sβ2M < 3.5 mg/L), patients were further divided into two sub-groups based on the expressions of 176 lncRNAs using k-means clustering (k = 2). The Kaplan–Meier curves of the two sub-groups were significantly different with p value < 0.0001 by log-rank test (Figure 4(a)). Similar result was observed in high-risk group with Sβ2M ⩾ 3.5 mg/L (Figure 4(b)).

Survival analysis of MM patients grouped by Sβ2M, ALB, and LDH levels in serum: (a) Kaplan–Meier survival curve of patients with low Sβ2M level (<3.5 mg/L), (b) Kaplan–Meier survival curve of patients with high Sβ2M level (⩾3.5 mg/L), (c) Kaplan–Meier survival curve of patients with high ALB level (⩾3.5 g/L), (d) Kaplan–Meier survival curve of patients with low ALB level (<3.5 g/L), (e) Kaplan–Meier survival curve of patients with high LDH level (>190 U/L), and (f) Kaplan–Meier survival curve of patients with low LDH level (<190 U/L).
Then, 558 patients in GSE20480 were divided into two groups based on the concentration of ALB in serum. In all, 481 patients with high level of ALB (ALB ⩾ 35 g/L) were clustered into two sub-groups with significant difference based on the expression (Figure 4(c)). Similar result was observed in low level of ALB (Figure 4(d)).
According to LDH level in serum, 558 patients were separated into two groups, herein, 168 were in high-level group (LDH > 190 U/L). The two groups were both sub-grouped using k-means clustering analysis with k = 2. Figure 4(e) displays significant difference between the sub-groups of high LDH level group, and Figure 4(f) shows that of low LDH level group.
GSEA
In GSEA of MM patients in GSE20480 dataset, many tumor-associated pathways were identified relating to survival of MM patients, including cell cycle (Figure 5(a)), focal adhesion (Figure 5(b)), and G2-M checkpoint (Figure 5(c)). It was obvious that pathways of cell cycle and focal adhesion changed significantly in MM patients with poor prognosis.

Biological pathway associated with lncRNA: (a) pathways related to cell cycle, (b) pathways related to focal adhesion, and (c) pathways related to G2-M checkpoint.
Discussion
MM is a malignant tumor characterized by wide clinical spectrum32,33 and pathophysiologic heterogeneities leading to fatal outcomes. The survival time of patients with MM ranges significantly from few weeks to more than 10 years. 32 Despite the remarkable improvements in treatment and patient care, 34 the 5-year survival rate is still nearly 40%, making MM to remain as an incurable disease. Due to the obvious differences in survival of MM patients, stratification of patient prognosis and accurate treatment are essential to improve the prognosis of patients. Recently, the prognosis of MM is stratified, mainly based on the levels of Sβ2M, LDH, ALB, and other protein. But considering the volatile secretion ability of diverse types of cells, the relationship between the concentration of these secretory proteins and malignant degree of tumor is not completely proportional. Therefore, these clinically relevant prognostic factors cannot truly reflect the prognosis of MM patients.
LncRNAs emerge as important regulatory molecules in tumor suppressor and oncogenic pathways. 35 Many studies led to preliminary hypotheses about the involvement of lncRNAs in diverse biological processes, such as stem-cell pluripotency and cell-cycle regulation.9,36 In particular, previous studies had observed a group of lncRNAs that are strongly associated with the p53 transcriptional pathway which is an important tumor-suppressor gene involved in maintaining genomic integrity. 37 However, studies on the application of lncRNA to predict the survival of MM patients were in deficiency. In this study, we analyzed the correlation between lncRNA expression and prognosis of patients with MM using a GEO database with a large number of patients’ data. The expressions of 176 lncRNAs were significantly correlated with the survival of MM patients. Subsequently, k-means clustering (k = 2) and Kaplan–Meier analysis found a statistically significant difference in survival between the two groups of patients sorted by the 176 lncRNAs. The same results were observed in an independent testing dataset GSE57317. Similar approaches on improving prognosis prediction using lncRNA have been studied in many cancers;38–42 however, few in MM. Top 20 out of 176 lncRNAs were most significantly expressed associated with survival of MM patients. Among these identified 20 lncRNAs, the top 3 lncRNA RP1-286D6.1, AC008875.2, and MTMR9L deserve more attention and analysis. In a recent study, RP1 (MAPRE2) was identified in a proteomic screen of pancreatic cell lines and that the high expression of RP1 mRNA was associated with poor outcome and prognosis. 43 The expression of lncRNA RP1-286D6.1, however, is identified as differentially expressed in MM for the first time. As to AC008875.2, there is a dearth of study reporting its association with any cancer. MTMR9L was suggested as novel protein phosphatases in dog. 44 Previously, this protein has not been found in human, mouse, rat, and cow. The GNASAS amplicon, part of the GNAS DMR2, was studied locating at the proximal promoter of this imprinted RNA antisense transcript of GNAS, 45 overlapping the binding site of several transcription factors according to ENCODE 46 CHIPseq data. RP1-286D6.1 and AC008875.2 were newly identified lncRNAs relating to MM.
Moreover, as to RP11-445H22.2, a similar lncRNA RP11-445H22.4 was suggested as biomarker for the diagnoses of breast cancer patients. 47 In that report, levels of serum lncRNA and the clinicopathological factors of these patients were further analyzed. Similarly, we also conducted a stratified analysis of the same pathological data to assure that lncRNA expression can predict the patient’s survival independent of other pathological characteristics of MM. Fortunately, we found that the concentrations of Sβ2M, LDH, and ALB were also significantly correlated with the overall survival of patients with MM. At the same time, GSEA of the identified lncRNAs was performed to further explore by which biological pathway that these lncRNAs influence the prognosis of MM patient. It was found that pathways relating to the cell cycle, focal adhesion, and G2-M checkpoint underwent significant changes in patients with poor prognosis. It has been reported that MM was characterized by overexpression of CCND1 or CCND2 influencing G1-phase cell-cycle progression to arrest in S/G2/M. 48 And it was found that Asiatic acid inhibits cell proliferation through regulating the expression of focal adhesion kinase in MM cells. We surmise that phenotypes promoting cell proliferation and inhibiting cell adhesion account for progress of MM. And further studies on the associating genes influencing these pathways should be screened.
Overall, a total of 176 lncRNAs were identified as significantly related to the survival of MM patients in this study. Meanwhile, these lncRNAs were applied to predict the survival of patients in dataset GSE24080 and GSE57317. These lncRNAs, especially RP1-286D6.1, AC008875.2, MTMR9L, AC069360.2, and AL512791.1, were potential biomarkers to evaluate the prognosis of patients with MM. However, the molecular mechanism of many pathways associated to the development of MM in which these lncRNAs participated needs further study.
Footnotes
Acknowledgements
A.-X.H. and Z.-Y.H. are co-first authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.