
Abstract
Astrocytomas are the most common primary brain tumors. They are very resistant to therapies and usually progress rapidly to high-grade lesions. Here, we investigated the potential role of DNA repair genes in astrocytoma progression and resistance. To this aim, we performed a polymerase chain reaction array-based analysis focused on DNA repair genes and searched for correlations between expression patters and survival prognoses. We found 19 genes significantly altered. Combining these genes in all possible arrangements, we found 421 expression signatures strongly associated with poor survival. Importantly, five genes (DDB2, EXO1, NEIL3, BRCA2, and BRIP1) were independently correlated with worse prognoses, revealing single-gene signatures. Moreover, silencing of EXO1, which is remarkably overexpressed, promoted faster restoration of double-strand breaks, while NEIL3 knockdown, also highly overexpressed, caused an increment in DNA damage and cell death after irradiation of glioblastoma cells. These results disclose the importance of DNA repair pathways for the maintenance of genomic stability of high-grade astrocytomas and suggest that EXO1 and NEIL3 overexpression confers more efficiency for double-strand break repair and resistance to reactive oxygen species, respectively. Thereby, we highlight these two genes as potentially related with tumor aggressiveness and promising candidates as novel therapeutic targets.
Introduction
Astrocytomas comprise the most common and malignant type of primary brain tumors affecting adults.1,2 The World Health Organization (WHO) classifies astrocytomas into four grades: pilocytic astrocytomas (grade I), which appear mainly in children, present slow proliferation rates, and can be cured by surgical resection; diffuse astrocytomas (grade II), characterized by moderate cellular proliferation, the presence of pleomorphic cells, and infiltrative capacity, which compromises complete resection and favors malignant progression; anaplastic astrocytomas (grade III), depicted by high cellularity, occurrence of extensive atypia, and the presence of mitotic figures; and glioblastoma multiform (GBM, grade IV), the most malignant and frequent type of astrocytoma, characterized by extremely increased mitotic activity, pronounced angiogenesis, the presence of pseudopalisading necrosis, and proliferative ratios from 3 to 5 fold higher than grade III tumors.1,3,4
The survival of patients diagnosed with GBM is usually 14 months, and less than 5% of them live through more than 3 years after diagnosis. This dismal prognosis is related with the intense mitotic activity and resistance of GBM cells to genotoxic treatments, which normally allows recurrence.2,5,6 Despite the impressive progress on research technologies and the intense efforts directed to GBM characterization, better diagnosis or advances on alternative therapies still seem remote. An increasing amount of genome scale profiling studies have recently provided a huge amount of data regarding genome structure and methylation and global patterns of gene expression of hundreds of GBM cases.7–11 Global analysis of genome structure and activity of currently available data confirmed previously reported genes as significantly mutated in GBM, namely, PTEN, TP53, EGFR, PIK3CA, PIK3R1, NF1, RB1, IDH1, and PDGFRA, and identified 61 additional genes. Furthermore, expression data analysis permitted a more refined classification of GBMs into four categories: proneural, neural, classical, and mesenchymal, which respond differently to treatment protocols.11,12 It was also demonstrated that a subset of proneural GBMs exhibits a hypermethylated phenotype of CpG islands (G-CIMP) associated with conspicuous better survivals, while non-G-CIMP proneural and not mesenchymal GBMs tended to show less-favorable outcomes.11,13 These patients usually present mutations in the IDH1 gene,12,13 which were independently associated with lower proliferation rates and better clinical outcomes in a cohort from China. 14
In this scenario, several molecular biomarkers have been independently associated with malignancy and survival prognosis of GBM patients, such as the MGMT gene. Methylation of the MGMT promoter is consistently correlated with longer overall survivals and can be used as a response predictor for patients under treatment with alkylating agents.11,15,16 Additionally, we have demonstrated that the overexpression of HJURP, a novel protein involved in centromeric chromatin assembly and DNA repair,17–19 is an independent factor of survival prediction for astrocytoma patients. 20 HJURP overexpression was also previously included in a four-gene signature associated with poor clinical outcome of high-grade gliomas. 21
Once DNA repair genes have exhibited this robust correlation with astrocytoma prognosis and therapy response, we decided to explore the different pathways involved in DNA repair signaling and execution during astrocytoma progression. We found 19 DNA repair genes with expression significantly altered in different-grade astrocytomas. Among them, seven genes (EXO1, NEIL3, RAD54L, XRCC3, BRCA2, BRIP1, and APEX2) showed remarkable high levels in GBM. Moreover, combining these 19 genes in all possible arrangements, we found 421 expression signatures strongly associated with poor survival. Importantly, five genes (DDB2, EXO1, NEIL3, BRCA2, and BRIP1) were independently correlated with survival, revealing single-gene signatures predictive of worse prognoses. Additionally, functional studies suggested that EXO1 and NEIL3 overexpression confers more accuracy for the repair of double-strand breaks (DSBs) and resistance to reactive oxygen species (ROS) in GBM cells, respectively.
Methods
Ethics statement
All patients involved provided a written consent for the use of tissue samples for research proposals. The consent form and research investigation were approved by the Ethics Committee of the Faculty of Medicine of Ribeirão Preto, University of São Paulo (HCRP 7645/99). Clinical procedures were conducted according to the principles of the Declaration of Helsinki.
Tissue samples and cell lines
Glioma samples were obtained from 55 patients submitted to surgical resection for tumor removal at the Clinical Hospital of the Faculty of Medicine of Ribeirão Preto, University of São Paulo. The set analyzed comprised 06 grade II and 06 grade III astrocytomas and 42 GBMs. Tumor grade was determined according to WHO criteria. 22 A total of 14 non-neoplastic white matter samples were obtained from patients undergoing amygdalohippocampectomy for drug-resistant epilepsy treatment at the same Hospital. In the polymerase chain reaction (PCR) array technique, 15 neoplastic samples (5 of each grade) and 5 non-neoplastic white matter samples were used. For quantitative reverse transcription polymerase chain reaction (qRT-PCR), we utilized 55 neoplastic (6 ASTII, 7 ASTIII, and 42 GBM) and 9 non-neoplastic white matter samples. Tumors and non-neoplastic samples were quickly frozen in liquid nitrogen immediately after surgical resection. Tumor fragments were microdissected to remove necrosis and non-neoplastic tissue and were kept in freezer at −80 °C until RNA extraction. Glioblastoma cell lines utilized, T98G and U138MG, were kindly provided by Professor Elza Tiemi Sakamoto Hojo, FFCLRP-USP, in 2012. Both cell lines were originally obtained from the American Type Culture Collection (ATCC).
Cell culture and transfection
T98G and U138MG cell lines were cultivated in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) with 10% of fetal bovine serum following standard protocols. For knockdown experiments, cells were transfected with synthetic double-stranded RNA oligonucleotides (short interfering RNAs (siRNAs); Invitrogen) and RNAiMax Lipofectamine (Invitrogen), following the manufacturer’s instructions. Oligonucleotide sequences are available upon request. Silencing of EXO1 and NEIL3 was confirmed by quantitative RT-PCR (Figure S1).
RNA extraction, PCR array, and quantitative RT-PCR
Total RNA from tissue samples was isolated using TRIzol Reagent (Invitrogen) following the manufacturer’s instructions with an additional phenol/chloroform extraction to improve protein exclusion. Purity and integrity of RNA were evaluated as previously described. 23 Complementary DNA (cDNA) synthesis was performed with High Capacity Kit (Applied Biosystems) after treatment with DNAse I (Invitrogen) in the presence of RNAse inhibitor (RNAseOUT; Invitrogen), according to manufacturer’s recommendations. PCR array analysis was carried out using the DNA Repair ChampionChIP™ PCR Array (SABiosciences), using Power SYBR Green PCR Master Mix reagent (Applied Biosystems). Data obtained were processed with the software available online (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php). qPCR reactions were conducted using the Power SYBR Green PCR Master Mix reagent and primers designed to seven selected DNA repair genes and for the HPRT gene, a control of constitutive expression previously demonstrated as adequate to this model. 23 Oligonucleotide sequences are available upon request. Reactions were performed in the 7500 Real-Time PCR System (Applied Biosystems), and the analyses were based on the 2−ΔΔCT equation. 24 Statistical analysis was performed with the non-parametric two-tailed test of Mann–Whitney (analysis of variance (ANOVA)) using the software GraphPad Prism 5.0.
Identification of correlations with survival prognosis
We developed an in silico strategy to identify expression signatures correlated with patient prognosis. Briefly, we utilized three datasets from The Cancer Genome Atlas (TCGA) containing RNA-seq count (RSEM) and clinical data, downloaded on 23 March 2015. Expression signatures containing all possible combinations of the 19 differentially expressed genes were generated. We computed the first principal component (PC1) of each signature and then divided the cohort into two groups according to a cutoff established by receiver operating characteristic (ROC) curve analysis (minimum area under the curve (AUC) of 0.7). The essential information used on Cox regression 25 was patient age, sex, tumor grade, and expression status, namely, carriers and non-carriers of signatures that were defined according to the cutoff. Patient age was stratified into two groups, <30 years and >30 years. MGMT methylation status was not available in the public datasets used (http://www.cbioportal.org/). Signatures considered independent parameters of prognosis correlation on Cox regression (p ≤ 0.001) were subsequently submitted to Kaplan–Meier analysis. Signatures with significant (p ≤ 0.001) predictive value of prognosis in both analyses are shown here.
Cell cycle, apoptosis and cell death, DNA damage, and proliferation assays
Cell-cycle analysis was performed through DNA content evaluation following standard protocols, and the data obtained were analyzed with the ModFit LT 3.3 software. Early apoptosis and effective cell death were measured with annexin V (Invitrogen) and propidium iodide labeling, respectively, followed by analysis in flow cytometer (FACSCanto; BD Biosciences). To evaluate the DSB amounts, cells were labeled with anti-γH2AX antibody (ABCAM), following the manufacturer’s instructions, and analyzed in flow cytometer (FACSCanto). The proliferation was evaluated with crystal violet staining followed by absorbance measurement. Results of all functional analysis are the average of three independent experiments.
Results
DNA repair genes presented altered expression in astrocytomas
In order to investigate the role of DNA repair genes in astrocytoma progression, we evaluated the expression of 84 genes involved in DNA damage signaling and repair, in a cohort of 15 different-grade astrocytomas through PCR array. Among them, we found 19 genes presenting expression significantly altered with large heterogeneity among astrocytoma grades and even between samples of the same tumor group (data not shown). In diffuse astrocytoma group (grade II), 14 genes showed increased expression, with average fold changes varying from 4.1 to 10.2 when compared with non-tumoral white matter. In anaplastic astrocytoma (grade III), 4 genes were down regulated and 7 genes were overexpressed, with fold change ranges extending from 0.005 to 0.17 and from 4.03 to 15.7, respectively. In GBM (grade IV), 7 genes were remarkably overexpressed varying from 4.1 to 20.9 fold (Table 1). These data revealed a meaningful amount of genes with altered expression and also suggested a correlation between fold change levels and tumor progression.
Average fold changes of significantly altered DNA repair genes in different-grade astrocytomas.

NA: not significantly altered.
The seven genes altered in the highest grade astrocytoma, namely, APEX1, BRAC2, BRIP1, EXO1, NEIL3, RAD54L, and XRCC2, were chosen for validation analysis in an expanded panel of 55 clinical cases, also including astrocytoma samples of grades II, III, and IV. We confirmed the higher expression of these genes, which exhibited fold changes even greater than those observed primarily. Variation was not significant only for RAD54L in ASTII samples (Figure 1). Analyzing the number of cases per fold change range, we noticed that most samples were in the expression range from 2 to 10 fold for APEX1, RAD54L, and XRCC2; from 10 to 60 for BRCA2 and BRIP1; and over 60 for EXO1 and NEIL3 genes (Table 2). Remarkably, EXO1 and NEIL3 showed the most extreme fold change values, increasing more than a 100 fold in ASTIII and GBM samples. When the expression levels were compared among tumor groups, we observed significant differences only for NEIL3 between ASTII and GBM samples (Figure 1).

Expression levels of selected DNA repair genes in a cohort of 55 different-grade astrocytomas. Expression levels of the indicated genes were evaluated by quantitative RT-PCR in samples of normal white matter (WM, n = 9), diffuse astrocytoma (ASTII, n = 6), anaplastic astrocytoma (ASTIII, n = 7), and glioblastoma multiforme (GBM, n = 42). Boxes represent lower and upper quartiles of relative expression ranges with medians indicated. Whiskers represent the 10th and 90th percentiles. *p < 0.05, **p < 0.01, and ***p < 0.0001, when comparing each tumor grade with WM (Mann–Whitney U test). Graphics were plotted using the software GraphPad Prism 5.0.
Number of astrocytoma samples per fold change range when comparing tumor tissues with non-tumoral WM.

Expression signatures of DNA repair genes are correlated with patient survival
Pursuing for combinations of expression patterns correlated with patient’s survival, we performed a bioinformatics analysis to identify expression signatures of DNA repair genes associated with higher death risk. Initially, the 19 genes found as differentially expressed were combined to generate all feasible arrangements of variation (signatures). The 1,048,575 possible combinations, ranging from each individual gene to all 19 genes in a single signature, were searched in public expression databases and compared with patient’s survival. We found 3357 signatures significantly correlated with worse survival prognosis. Patients carrying these signatures presented hazard ratios (HRs) above 1.62. We established a cutoff of 5 genes per signature and HR ≥ 2 and selected 421 expression signatures strongly correlated with patient survival (Table S1). Among them, 5 occurred as signatures of unique genes. The overexpression of NEIL3, EXO1, BRCA2, and BRIP1 and the reduction of DDB2 were independently correlated with higher risk of patient’s death, with HR varying from 1.75 to 3.22 (Table 3). Additionally, DDB2 gene was present in 99.4% of signatures, revealing the importance of DDB2 downregulation for GBM development (Table S2), and when signatures were ranked by HR values, the first 67 signatures included DDB2 with HR varying from 4.73 to 3.22. Importantly, the last one in this ranking encloses DDB2 downregulation as the single alteration (Table S1), reinforcing the major importance of DDB2 reduction for astrocytoma progression. The altered expression of RAD54L, XRCC2, MSH6, LIG3, PNKP, FEN1, and MSH5, although did not correlate with prognosis independently, also promoted a meaningful HR (from 4.32 to 2.03) when coupled with DDB2 downregulation (Table 3). Overexpression of EXO1 and NEIL3, which exhibited independent association with poor prognosis, revealed even greater HRs when combined with DDB2 reduction, showing an increment in death risk from 2.13 and 2.15 to 4.73 and 4.44 fold, respectively. Accordingly, patients who carry these signatures have a prominent reduction in life expectancy (Figure 2). Due to these observations, we chose EXO1 and NEIL3 for functional analysis of gene silencing in GBM cell lines.
Hazard ratios of patients carrying signatures containing one or two genes.


Kaplan–Meier survival curves for selected gene signatures. Dotted curves denote patients with PC1 value (first principal component) above the cutoff (defined by ROC curve analysis). Continuous curves denote patients with PC1 value below the cutoff. Log-rank tests resulted in p < 0.001.
Knockdown of EXO1 and NEIL3 affects cell-cycle dynamics and DNA repair activity of GBM cells
To further investigate the potential roles of EXO1 and NEIL3 overexpression in the resistance of GBM cells to DNA damage induction, we silenced these genes in T98G and U138MG cell lines, which express high levels of both genes (data not shown), and evaluated response to ionizing radiation (IR). As expected, IR promoted prominent changes in cell-cycle dynamics in the two cell lines analyzed, with reduction in the number of cells in G1 and increase in G2/M phases (Figure S2). However, when cells were irradiated and simultaneously silenced for EXO1, we did not observe additional alterations in cell-cycle dynamics. Interestingly, the isolated knockdown of EXO1 promoted a slight augment in the number of cells in G1 and a reduction in S phase for T98G cell line (Figure S2A), while U138MG was not significantly affected (Figure S2B; Figure 3(a)). Additionally, we observed that EXO1 knockdown supported a faster DNA DSB restoration after IR exposure in T98G (Figure S3A) but not in U138MG cells (Figure S3B). T98G cells silenced for EXO1 showed approximately 30% and 15% reduction in the amounts of DSBs 10 min and 3 h after irradiation, respectively; and 6 h after IR exposure, there was no significant difference from cells treated with siCtrl RNAs (Figure 3(b)). Accordingly, T98G cells silenced for EXO1 presented higher proliferation rates (Figure 3(c)) and slightly reduced indexes of apoptosis (Figure 4), when compared to cells treated with scrambled siRNAs.

Cell-cycle dynamics, DNA damage, and proliferation of GBM cells after EXO1 or NEIL3 silencing. Cells were treated or untreated with 14 Gy of IR (RS 2000 X-ray irradiator, 25 mA and 160 kVp) 3 days after transfection with siRNAs directed to EXO1 or NEIL3. (a) At the fifth day after transfections, cells were fixed and stained with propidium iodide and cell-cycle distribution was evaluated by flow cytometry. (b) Cells were fixed at the fifth day after transfection, labeled with anti-γH2AX antibody (ABCAM), and analyzed by flow cytometry analysis. (c) Proliferation rates were estimated by staining with crystal violet at each 24 h after transfection during 5 days. The graphs were plotted with the software “GraphPad Prism 5.0” (*p < 0.05, **p < 0.001, and ***p < 0.0001).
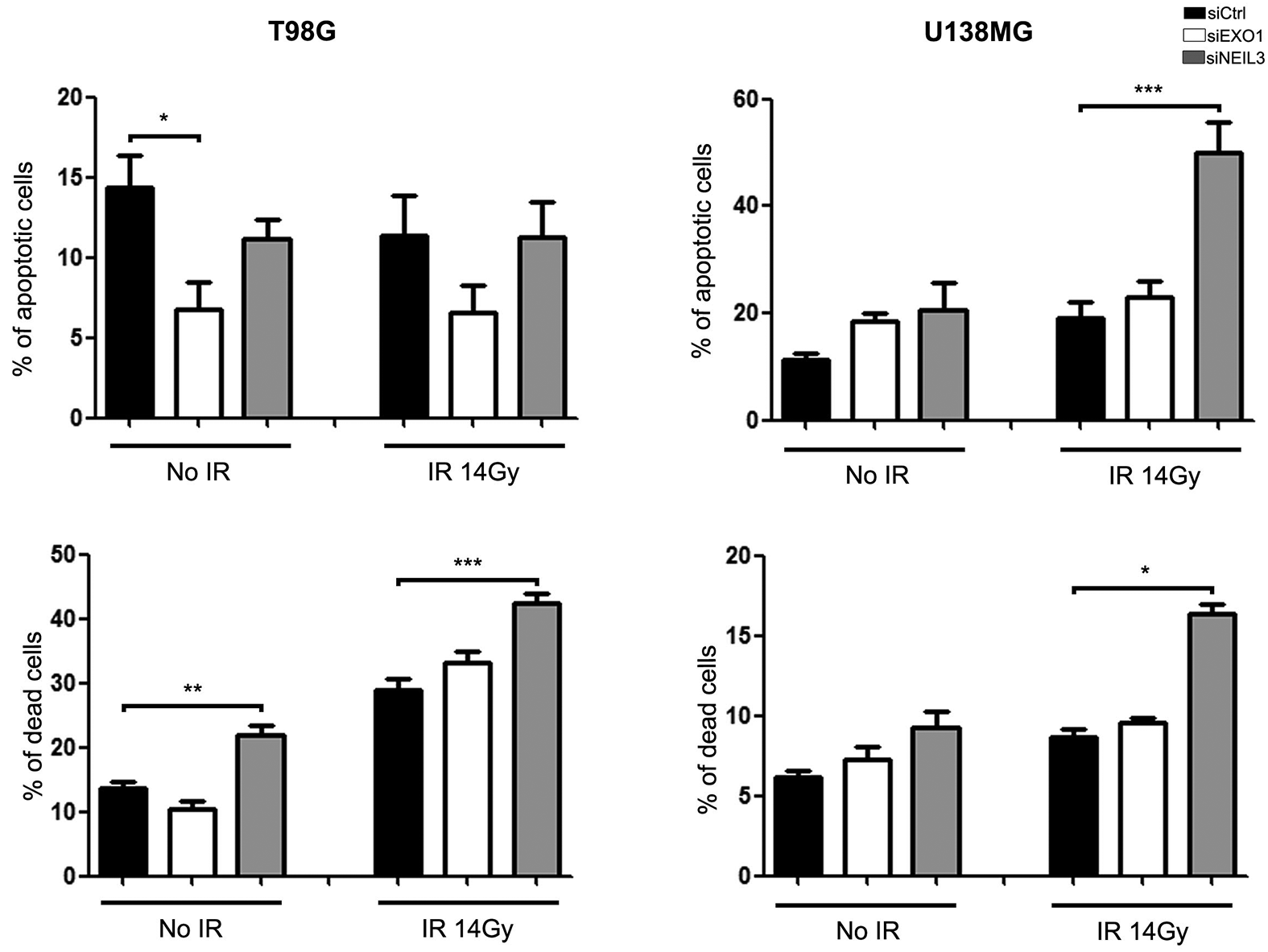
Quantification of apoptosis and effective cell death in GBM cells after silencing EXO1 or NEIL3. Cells were treated or untreated with 14 Gy of IR (RS 2000 X-ray irradiator, 25 mA and 160 kVp) 3 days after transfection with siRNAs directed to EXO1 or NEIL3. Numbers of apoptotic or dead cells were estimated by flow cytometry detection of annexin V and propidium iodide at 48 h after irradiation. Graphs were plotted with the software GraphPad Prism 5.0 (*p < 0.05, **p < 0.001, and ***p < 0.0001).
The isolated knockdown of NEIL3 did not impact cell-cycle dynamics of T98G and U138MG cells (Figure S2). Otherwise, silencing of NEIL3 combined with irradiation affected both cell lines. We observed a reduction in the number of U138MG cells in G1 and an increase in G2/M phases (Figure S2B), while the number of T98G cells were diminished in S and augmented in G2/M phases (Figure S2A and Figure 3(a)). We also observed pronounced higher levels of DSB after IR treatment for T98G cells silenced for NEIL3 at all times evaluated (Figure S3A and Figure 3(b)), while for U138MG cells a discreet increase was observed only after 6 h of exposure (Figure S4). Consistently, we detected augmented levels of cell death in both cells after irradiation and NEIL3 knockdown (Figure 4 and Figure S5). Apoptosis rates, conversely, were greater only in U138MG, in accordance with the wild-type genetic status of TP53 gene, which is mutated in T98G cells. These results reveal that cell death activation occurs by a mechanism other than apoptosis in these cells. We also observed that the unique reduction of NEIL3 promoted an increment in cell death for T98G cell line (Figure 4), correlating with the notable accumulation of DSBs after irradiation and NEL3 silencing (Figure 3(b)).
Discussion
In this work, we identified 19 DNA repair genes with altered expression in different-grade astrocytomas, the majority of them showed overexpression and 4 were detected at reduced levels in tumor tissues. Interestingly, we observed a fewer number of altered genes in GBMs when compared to lower grade astrocytomas and a tendency to higher expression levels in more aggressive tumors. These data indicate that the upregulation of selected DNA repair genes develops in parallel with the cumulative genomic instability observed during tumor progression. Supporting this statement, it was reported that DNA damage response (DDR) activation and amounts of DSBs are reduced in higher grade gliomas.26,27 Recently, Turner et al. 28 have shown a reliable enrichment in DNA repair activity of non-homologous end joining (NHEJ) and homologous recombination pathways during glioma progression mediated by Akt3 activation.
Additionally, we observed a strong correlation between the expression of DNA repair genes we found altered and patient survival prognoses. Among them, five genes (DDB2, NEIL3, EXO1, BRCA2, and BRIP1) showed an independent correlation with poor prognoses, defining signatures of single genes. DDB2 was detected at lower levels, while the other four showed higher expression in astrocytomas. Importantly, DDB2 downregulation presented the highest HR (3.22) among the single-gene signatures and was present in 99.4% of signatures. DDB2, which is involved in the nucleotide excision repair (NER) pathway, is a remodeling enzyme of damaged DNA that acts in initial steps of NER. DDB2-DDB1 complex recognizes DNA damage and facilitates the recruitment of XPC to lesion sites.29,30 Thus, downregulation of DDB2 possibly promotes accumulation of DNA damage allowing a bypass in the DDR triggered by replicative stress. Interestingly, the reduction in DDB2 expression was correlated with poor outcomes of ovarian cancer patients, and its overexpression suppressed the ability of ovarian cancer cells to recapitulate tumors in athymic nude mice by reducing the cancer stem cell population. 31 DDB2 was also postulated as a suppressor of epithelial-to-mesenchymal transition (EMT) in colon cancer, 32 suggesting that when DDB2 is reduced tumor cells undergo EMT and acquire a more aggressive behavior. These results corroborate our findings that correlated DDB2 reduction with more aggressive phenotypes of GBM.
Curiously, the overexpression of the other four single-gene signatures was also associated with higher risk of death. Overexpression of EXO1 and NEIL3, which are enzymes involved in DNA repair execution, showed an increased HR when combined with downregulation of DDB2. Patients carrying the DDB2_EXO1 or DDB2_NEIL3 signatures presented a faster progression of the disease and an augmented risk of early death of 4.73 and 4.44 fold, respectively. The overexpression of RAD54L and XRCC2, which are involved in the homologous recombination repair pathway and did not correlate with prognosis independently, also promoted a meaningful HR when coupled with DDB2 of 4.32 and 3.85, respectively. Altogether, these results suggest that permissiveness for mutation accumulation allied to higher competence in DNA repair activity can favor tumor development, by preventing collapsing levels of genomic instability that could cause cell death. Bartek et al. 33 have recently proposed a dual role for the ATR-Chk1 pathway, tumor suppressive and tumor promoting in early and advanced stages of cancer, respectively, which supports this hypothesis.
Due to the remarkable impact of EXO1 and NEIL3 overexpression for patient prognosis, we evaluated the effects of silencing these genes in cell-cycle dynamics, DSB repair activity, proliferation capacity, and cell death indexes of T98G and U138MG cell lines after exposure to IR. When EXO1 was silenced, T98G cells repaired DNA damage faster and presented higher levels of proliferation, indicating that EXO1 reduction favored DSB restoration. EXO1 is a 5′ to 3′ exonuclease that resects DSB with blunt ends generating a single-strand tail that is necessary for the invasion of the intact double-strand DNA used as template for homologous recombination repair. 34 Thus, T98G cells presenting reduced levels of EXO1 possibly directs the repair activity of DSB for the NHEJ pathway, which is an error-prone mechanism that promotes a faster reparation of these DNA lesions. In agreement with our hypothesis, Tomimatsu et al. 35 have shown that EXO1-mediated DNA resection is a pivotal event in repair pathway choice between homologous recombination and NHEJ. Moreover, it was also demonstrated that NHEJ efficiency was not affected in a Saccharomyces cerevisiae strain mutant for EXO1, although repair accuracy was reduced. 36 In contrary, U138MG cells were not affected by EXO1 knockdown. This difference is probably associated with the genetic background and proliferation indexes of these cells, once T98G cell line is mutant for TP53 37 and present shorter doubling (data not shown).
NEIL3 knockdown affected both cell lines analyzed similarly. We observed cell-cycle arrest in G2/M phase, and higher percentage of damage and cell death when cells were silenced for NEIL3 and irradiated, demonstrating that NEIL3 reduction sensitizes GBM cells to IR. NEIL3 is a glycosidase that excises oxidative damage of bases generating apurinic/apyrimidinic sites that are recognized and converted into DNA single-strand breaks by the endonuclease APEX2. 38 Coherent with a potential function in the handling of ROS, generated by replicative stress, it was reported that NEIL3 is upregulated in S phase of cell cycle and acts preferentially on single-stranded DNA.39,40 Accordingly, Takao et al. 38 demonstrated that an Escherichia coli strain double mutated for Nth and Nei presents higher sensibility to ROS, and when Neil3 is overexpressed, the resistance was partially recovered. Similarly, Rolseth et al. 41 demonstrated that the primary mouse embryonic fibroblast (MEFs), obtained from Neil3 knockout mouse, presented higher sensibility to the genotoxic agents cisplatin and paraquat, when compared to MEFs with normal expression of Neil3. Once secondary DNA damage inflicted from IR is promoted by ROS, our data suggest that similar to Takao et al. 38 and Rolseth et al., 41 NEIL3 could play an important role in the resistance of GBM cells to this type of genotoxic stress.
In this study, we identified important expression signatures of DNA repair genes strongly correlated with astrocytoma progression. Therefore, genes enclosed in these signatures could represent new biomarkers for the prediction of disease prognosis and are potential targets for treatment. Particularly, we highlight the overexpression of EXO1 and NEIL3, which were independently associated with more than 2 fold increment in death risk for astrocytoma patients. Functional studies corroborated the relevance of EXO1 and NEIL3 overexpression for DNA repair activity and resistance to IR, emphasizing the importance of these genes also as promising candidates for the development of adjuvant therapies. Further studies using more refined approaches are required to functionally characterize the exact roles of these genes in astrocytoma biology.
Footnotes
Acknowledgements
The authors thank Silvia Regina Andrade Nascimento and Benedita Oliveira Souza for technical assistance. The authors also thank Fabiana Rossetto de Morais who supported all flow cytometry analyses.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
V.V. was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (grant no. 2013/13465-1) and Programa de Apoio ao Desenvolvimento Científico (grant no. 204/2012) from Faculty of Pharmaceutical Sciences of Araraquara. W.A.d.S., C.G.C., and V.V. were supported by Center for Cell-Based Therapy (CEPID/FAPESP; grant no. 2013/08135-2). J.F.d.S., R.B.S., and L.F.M.D.C. received fellowships from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.