
Abstract
The CINdex Bioconductor package addresses an important area of high-throughput genomic analysis. It calculates the chromosome instability (CIN) index, a novel measurement that quantitatively characterizes genome-wide copy number alterations (CNAs) as a measure of CIN. The advantage of this package is an ability to compare CIN index values between several groups for patients (case and control groups), which is a typical use case in translational research. The differentially changed cytobands or chromosomes can then be linked to genes located in the affected genomic regions, as well as pathways. This enables in-depth systems biology–based network analysis and assessment of the impact of CNA on various biological processes or clinical outcomes. This package was successfully applied to analysis of DNA copy number data in colorectal cancer as a part of multi-omics integrative study as well as for analysis of several other cancer types. The source code, along with an end-to-end tutorial, and example data are freely available in Bioconductor at http://bioconductor.org/packages/CINdex/.
Keywords
Introduction
DNA copy number change is an important form of structural variation in human genomes. Variations in copy number of germline cells are commonly referred to as copy number variations (CNVs), whereas changes in copy number arising from tumor tissue are commonly referred to as copy number alterations (CNAs).
Genomic instability is known to be a fundamental trait in the development of tumors, and most human tumors exhibit this instability in structural and numerical alterations: deletions, amplifications, inversions, or even losses and gains of whole chromosomes or chromosome arms. The chromosome instability indicated by these CNAs in the DNA has been associated with various events in the development or severity of tumors.
These DNA copy number changes can be measured by various technologies—including microarray-based comparative genome hybridization (arrayCGH) or genotyping arrays, and, more recently, high-resolution next-generation sequencing (NGS). 1
To mathematically and quantitatively describe these DNA CNAs, their genomic positions and ranges are first located. Each range, referred to as a segment, reflects a genomic region that has a similar genomic alteration profile. Each segment is assigned a numeric value that reflects the genomic instability in that segment. These segments are always unique to each patient. Such algorithms are referred to as segmentation algorithms.
Various methods have been proposed for copy number change detection. For CNV/CNA detection in a single profile, representative methods include Gain and Loss Analysis of DNA, 2 Circular Binary Segmentation (CBS), 3 and Hidden Markov Model (HMM). 4 Consensus CNV/CNA detection methods can be categorized into 1-stage and 2-stage approaches. A 2-stage approach, such as Genomic Identification of Significant Targets in Cancer (GISTIC), 5 involves a step of copy number change detection in individual profiles and a subsequent statistical analysis of commonly altered DNA regions. A 1-stage approach, such as the Bayesian Segmentation Approach (BSA), 6 can directly detect common CNV/CNA patterns shared by multiple signal profiles.
Bioconductor is an open-source, open-development repository of software packages built using the R programming language. 7 Bioconductor has several copy number segmentation algorithms including copynumber, 8 fastseg, 9 Vega, 10 SMAP, 11 and biomvRCNS. 12 There are several copy number segmentation algorithms outside of Bioconductor, including Fused Margin Regression (FMR) 13 and CBS. 14
One of the challenges with segmentation algorithms is that the segments are unique to each patient, which complicates a global analysis of the segment data in a group of patients.
We have created a Bioconductor package—CINdex—that can perform a global analysis of the segment data in a group of patients. The CINdex Bioconductor package accepts the segment information from any segmentation algorithm. It calculates a novel measure of genomic instability across a chromosome (referred to as Chromosome-CIN, Standard-CIN, or Regular-CIN) for a global view of genomic instability and across cytobands (referred to as Cytobands CIN) for a higher resolution of genomic instability.
The advantage of the CINdex package is that the CIN values are calculated at chromosome level and cytoband level, which are standard regions across the entire human genome. It hence allows comparison of chromosome instability values between several groups for patients (control vs case), which is a typical use case in translational research. In addition, the package also allows further downstream systems biology analysis by connecting the differentially changed cytobands or chromosomes to genes and pathways.
A simplified version of the CINdex algorithm that shows overall instability (ie, both losses and gains represented as an instability without alluding to a loss or gain) has been integrated into The Georgetown Database of Cancer (G-DOC) Web portal and made available for users for free as part of its toolkit at https://gdoc.georgetown.edu.15,16
Methods
The framework for the CIN analysis pipeline is shown in Figure 1A. The DNA copy number data are first obtained from one of many sources such as NCBI’s (National Center for Biotechnology Information) Gene Expression Omnibus, The Cancer Genome Atlas, or G-DOC, or user’s own data.
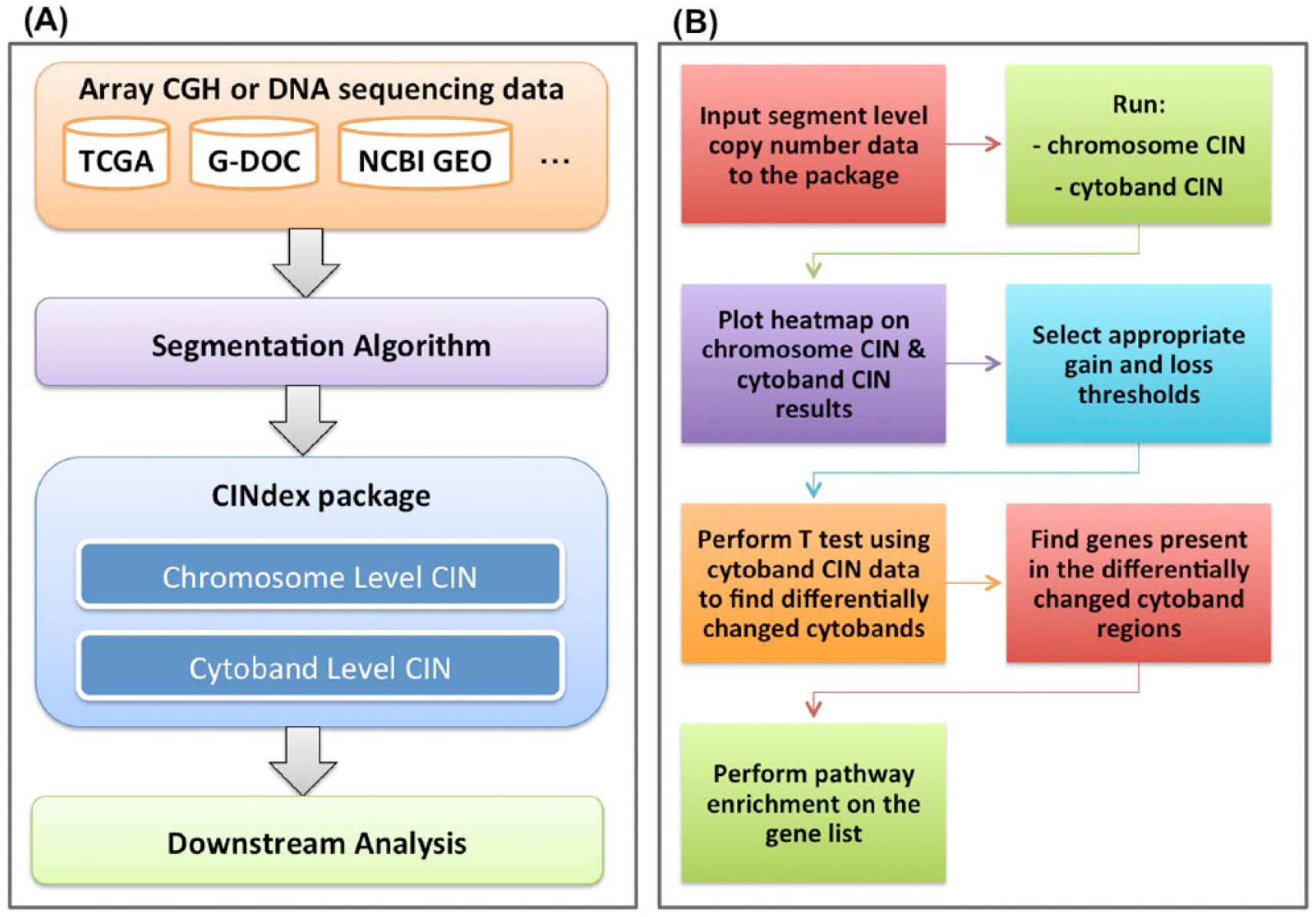
(A) The framework of the chromosome instability (CIN) analysis pipeline. (B) An end-to-end analysis using the CINdex package.
In this article, we demonstrate the working of the CINdex Bioconductor package using a portion of the data set from the work by Madhavan et al 17 consisting of 10 samples (5 relapse and 5 relapse-free). This data set is also available in G-DOC.15,16 This allowed us to reproduce the results from the paper and hence serve as a validation of the working of the package. This use case has been made into a tutorial and is available for download (as a vignette, Supplementary File 1) along with the package at http://bioconductor.org/packages/CINdex/. In addition to this tutorial, the package also includes another detailed tutorial on how to prepare input data for the package, along with downstream biological interpretation. The steps in brief on how to run an end-to-end analysis are shown in Figure 1B.
The input to CINdex package is segmentation data. It can accept segment data from any segmentation algorithm.
After the raw copy number data were obtained, we first applied a segmentation algorithm to obtain the segments and its values for each sample. For this use case, we used the FMR 13 as the segmentation method. We chose FMR as it had several advantages compared with other detection methods: (a) FMR is a unified computational model for detecting both copy number changes in a single profile and consensus copy number changes in population data; (b) FMR has higher sensitivity for low signal to noise ratio signal profiles compared with probabilistic model-based methods (eg, HMM) and thus would be more effective in detecting complex CNA patterns in tumor genomes; (c) FMR generates more accurate estimates of breakpoint loci compared with other regression-based methods18-20; and (d) FMR is empowered by a modified path algorithm 19 and hence is computationally efficient for detecting CNVs/CNAs in high-density microarray data.
For the purposes of reproducibility, efficiency, and scalability, input data files for the CINdex package were converted into standard compressed data structures. One such format is the GRanges class object from the GenomicRanges Bioconductor package. 21 The segment data, which can be large in size, are converted into this GRanges compressed data structure. Similarly, the platform annotation file and the genome reference files are also saved as GRanges objects. Detailed notes on how to convert input data into the GRanges object is provided along with the CINdex Bioconductor package in the file “How to prepare input data.pdf” (also as Supplementary File 2).
We would like to note that for this example use case, genome reference hg18 was used. This is because the analysis and results of the colon cancer data set 17 were performed using the hg18 reference genome. Our intent is to demonstrate the use of the CINdex package using the same data set as in the work by Madhavan et al 17 and reproduce the results from their work, 17 serving as a validation of our package. CINdex could be used with any reference version, as long as the same genome reference version used for segmentation is used for CINdex as well.
Once the input files (the segment data, the platform annotation file, and the genome reference file) were converted into the GRanges object, it was ready for the CINdex package. The CINdex package then calculated the chromosome instability index (CIN) values at the chromosome and cytoband levels.
Chromosome instability index is calculated by the following steps:
Given
the signal profile of chromosome
the segments generated by any segmentation method,
Steps are as follows:
Make gain/loss calls on the segments. A segment with mean signal intensity greater than
For each gain segment, its amplitude is the mean signal intensity;
Get the maximum gain amplitude
For each loss segment, convert its amplitude
Compute the chromosome-specific instability index
The same calculation is also done at the cytoband level to get cytoband-specific instability index.
The CIN values are calculated for several gain and loss thresholds. These thresholds define the cutoff values for a CIN value to be defined a gain or loss. By default, the following gain thresholds of 2.5, 2.25, and 2.10 and loss thresholds of 1.5, 1.75, and 1.90 are used to optimize visualization quality. Users can also input one or more of their own gain and loss thresholds. For each of these threshold settings, the algorithm calculates CIN values for gains, losses, and a combination of gains and losses (referred to as sum or overall CIN). For each threshold setting, CIN is also calculated with normalization (using a scaling factor based on median) and without normalization (no scaling factor used).
Using this algorithm, a CIN value was generated for each sample and for every chromosome and then collated into a matrix. Similarly, for each sample, the CIN values were generated for each cytoband and collated into another matrix. Hence, for every threshold setting, a chromosome-CIN matrix and a cytoband-CIN matrix are generated for gains, loss, and overall CIN. Example matrices for Chromosome-CIN and Cytoband-CIN are shown in Figure 2A and B, respectively.

Examples of (A) chromosome-CIN matrix—samples are columns and chromosomes are rows and (B) cytoband-CIN matrix—samples are columns and cytobands are rows. The red-colored matrix contains only amplifications, the blue-colored matrix shows only deletions, and the yellow-colored matrix contains the sum of both (also referred to as “overall CIN”). CIN indicates chromosome instability.
Once the CIN matrices are obtained, they can be visualized in the form of heatmaps. The purpose of generating CIN values for multiple thresholds is to allow the user to pick the appropriate gain and loss threshold/setting. The appropriate threshold is the one that shows the best contrast between 2 groups of interest.
Once the appropriate gain and loss thresholds were chosen, a T test was performed on the CIN values to compare the case (relapse) and control (relapse-free) groups. This produced a list of differentially changed cytobands (and/or chromosomes). It was interesting to see which genes belong to these significant cytoband regions. In the last step of the workflow, the gene symbols obtained in the previous step were used to perform pathway enrichment using the Reactome database within Bioconductor (using the ReactomePA Bioconductor package). 23
Our CINdex Bioconductor package has built-in functions that allow users to perform each of the abovementioned steps. This hence provides the user the ability to perform an end-to-end analysis, starting from DNA copy number data and all the way up to genes and pathways.
The CINdex algorithm is also available for omics data analysis inside the G-DOC platform.15,16 A simplified version of the algorithm has been implemented in G-DOC for easy usability and interpretation. This simplified version displays only overall CIN (sum of losses and gains), meaning both losses and gains are displayed as an “instability” without differentiating between a loss and a gain.
Results
In this article, we demonstrate an analysis of copy number data with CINdex Bioconductor package using a portion of the data set from Madhavan et al 17 consisting of 10 samples (5 relapse and 5 relapse-free).
Once the input files were converted into the GRanges object, it was ready for the CINdex package. Detailed notes on how to convert input data into the GRanges object are provided as Supplementary File 2. The CINdex algorithm calculated the CIN values at the chromosome and cytoband levels, and chromosome-CIN and cytoband-CIN matrices were generated.
Once the CIN matrices were obtained, they were visualized in the form of heatmaps. Figure 3 shows chromosome-CIN heatmaps generated for 3 gain and loss threshold combinations (with unnormalized setting): gain threshold of 2.1 and loss threshold of 1.9, gain threshold of 2.25 and loss threshold of 1.75, and gain threshold of 2.5 and loss threshold of 1.5. On comparing the 3 images, it was clear that the image from gain-loss threshold setting of 2.1 and 1.9 (Figure 3, top) was too dense, and the image from gain-loss threshold setting of 2.5 and 1.5 (Figure 3, bottom) was too sparse. This indicated that the setting with gain threshold of 2.25 and a loss threshold of 1.75 (Figure 3, middle) showed the ideal contrast between 2 groups of interest—relapse and relapse-free groups. As a future feature, we plan to automate the selection of the best threshold setting.

Chromosome-CIN heatmaps for gain and loss thresholds: 2.1 and 1.9 (top), 2.25 and 1.75 (middle), and 2.5 and 1.5 (bottom) showing amplifications (gains), deletions (losses), and overall (sum) CIN. CIN indicates chromosome instability.
Once the appropriate gain and loss thresholds were chosen, the CIN values can be used for downstream analysis and interpretation. As an example, the cytoband-level heatmap for chromosome 20 obtained from this data set analysis is shown in Figure 4. It was obtained using a gain threshold of 2.25 and a loss threshold of 1.75 with unnormalized setting. The nonrelapse group has more blue-colored bands compared with the relapse group. The blue color indicates deletions (losses).

Cytoband-CIN heatmap for chromosome 20. Blue color indicates genomic instability (losses). Black color indicates no genomic instability. CIN indicates chromosome instability.
A T test was performed on the CIN values to compare the case (relapse) and control (relapse-free) groups. Figure 5 is a heatmap showing statistically significant differentially changed cytobands between 2 groups (relapse and relapse-free). Among the top results are regions 4q, 16q, and 20q.

Heatmap showing statistically significant differentially changed cytobands between 2 groups.
Once we got the list of differentially changed cytobands, it was interesting to see which genes belong to these significant cytoband regions. This was performed using a built-in function in our package that allows to find genes present in the cytoband regions (Supplementary File 3).
In the last step of the workflow, the gene symbols obtained in the previous step were used to perform pathway enrichment using the Reactome database within Bioconductor. 23 Figure 6 shows the top pathway results of pathway enrichment in the form of a bar plot. The enrichment analysis of cytobands affected by chromosome instability indicated enrichment of specific pathways and biological processes related to immune response. These results are consistent with findings obtained in the work by Madhavan et al 17 using multi-omics data including gene expression, exome deep sequencing, metabolomics, and microRNA.

Pathway enrichment using Reactome database. The colors in the bars represent P values.
All these steps were done using the CINdex package. This shows that this package allows assessing the impacts of CNAs on various biological events or clinical outcomes by studying the association of CIN indices with those events. Hence, the CINdex package enables a complete end-to-end analysis on a data set without having to digress and use another software/tool.
Discussion
CINdex package was successfully applied to analysis of DNA copy number data in colorectal cancer as a part of multi-omics integrative study providing new insights for predictive biomarkers of relapse for stage II colorectal cancer, 17 as well as in multiple studies with a simplified version as part of the G-DOC platform.
The example shown in this use case was done on somatic structural alterations in tumor studies. It demonstrates the comprehensive systems biology application of the CINdex package—by connecting the differently changed cytobands to genes and pathways, it allows users to gain insights on the development or the severity of tumors in terms of clinical outcome.
In Figure 4, we can see more genomic instability in the relapse-free group compared with relapse groups. Higher risk groups (such as relapse) are known to have frequent fractional CNAs, whereas whole chromosomal arm CNAs are seen more in lower risk groups. 24 It is known that frequent fractional aberrations in DNA cause mutations and regulatory changes. 25 This is consistent with our findings where we see almost the entire chromosome lost in the relapse-free group compared with the relapse group.
The T test results list regions 4q, 16q, and 20q as the top significant differently changed cytoband regions. Chromosome losses in 4q region have been previously associated with local recurrence in colon cancers after surgical resection.26,27 The 16q region is the second most frequent target of loss of heterozygosity (LOH) in breast cancer. 28 High frequency of LOH has been associated with metastasis.29,30 The 16q region is also frequently methylated in lymphomas 31 —the CpG island methylator phenotype pathway, associated with methylation changes, is one of the known pathways of genomic instability in colon cancer. 30
In the heatmap shown in Figure 5, we see cytoband region 20q13.12 listed among the top statistically significant regions. This region is known to contain several oncogenes including MMP9, MYBL2, and UBE2C. MMP9 is known to facilitate metastasis by promoting matrix degradation and cell migration, MYBL2 is a transcription factor involved in cell cycle progression and antiapoptosis, and UBE2C is overexpressed in many different types of cancers and is associated cell cycle progression and tumor differentiation. 23
A simplified version of the CINdex algorithm has been implemented inside the G-DOC platform. The G-DOC system currently contains 8 studies (data sets) with copy number data that can be analyzed in conjunction with clinical or other omics data types. The main advantage of the CINdex algorithm is that it enables comparison chromosome instability values between several groups for patients (control vs case), a typical use case in translational research. Supplementary File 4 shows several of these comparisons performed using the CINdex algorithm inside the G-DOC platform.
The first figure in Supplementary File 4 (Figure S4A) shows a CIN heatmap from the NCI REMBARNDT study in G-DOC 16 comparing copy number data between 2 glioma types—astrocytoma (low-grade glioma) and glioblastoma (GBM, high-grade glioma). In the image, we see a higher level of chromosome instability in the astrocytoma group in the 8q region (indicated by the bright red colors). Aberrations in the 8q region in patients with astrocytoma are known in literature.32,33 In addition to 8q region, 7p and 10q regions were also unstable in GBM compared with astrocytoma. The 7p and 10q regions are known to be highly amplified in patients with GBM.34–37
The second figure in Supplementary File 4 (Figure S4B) shows a screenshot of T test results performed using the CRC_BROSENS_2010_01 study in G-DOC. The T test compared Cytoband-CIN data from patients who relapsed with patients who were relapse free (Disease-Free Survival = Event vs Disease-Free Survival = Censoring). The results showed that the most differentially changed cytoband regions are in chromosomes 3, 4, and 18. From the T test results, we can see that the most differentially changed cytoband regions include regions 3p, 4q, and 18q. Chromosome instability 4q has been previously associated with local recurrence in colon cancer 38 ; deletions in chromosome 3p have been associated with distant metastasis and poor survival in colorectal cancer. 39 The T test results include cytoband region 18q.21.2 among its top significant results, which contains the DCC gene where frequent LOH events in colon cancer occur. 40
The results of the CINdex algorithm applied using both Bioconductor and G-DOC are consistent with previously reported biological findings, which clearly demonstrates a utility of the CINdex package for fast and effective exploration of DNA copy number data.
The source code, along with an end-to-end tutorial, and example data are freely available in Bioconductor at http://bioconductor.org/packages/CINdex/. Supplementary File 5 shows a screenshot of the download statistics of the CINdex Bioconductor package as of May 30, 2017, showing more than 1600 downloads in total since its release.
Conclusions
CINdex package can analyze experimental CNV and CNA data generated by Affymetrix SNP 6.0 arrays or NGS technologies on studies with germline, structural, or somatic variations. It allows users to perform end-to-end analysis to assess the impacts of CNAs on various biological events or clinical outcomes.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
LS and YG designed the algorithm. LS wrote the R code. KB wrote the Bioconductor package. KB and YG wrote the manuscript. YW, YF, IS, and SM provided editorial comments. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication, authors have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. The external blind peer reviewers report no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.