
Abstract
Bladder cancer is the most common malignant tumor of urinary system, largely resulting from failure of repair of DNA damage to the environmental insults. The function of XPA in nucleotide excision repair pathway has been well documented. However, participation of XPA in the repair of DNA double-strand break remains unknown. Here, we reported that bladder cancer expressed low XPA levels compared to adjacent non-tumor bladder tissue, and this phenotype was closely associated with chromosomal aberrations. Moreover, downregulated XPA appeared to increase incidence of chromosome aberration. XPA reduction increased cell viability of a bladder cancer cell line RT4, while XPA re-expression decreased the cell viability of RT4 cells. Since high mutation frequency is the basis of mutations of oncogenes and anti-oncogenes, and may be the essence of bladder cancer susceptibility, our study suggests that downregulated XPA may promote carcinogenesis of bladder cancer via impairment of DNA repair.
Introduction
Bladder cancer (BC) is the most common malignant tumor that originates from urinary system, and failure of DNA damage repair in response to environmental insults including chemical dyes, smoking, and chemical drugs causes mutation and tumorigenesis of BC.1–7 Therefore, the DNA repair pathways play important roles in maintaining genomic stability and fidelity. If the mechanism of DNA repair changed, the lesions result in persistent DNA damage, which eventually leads to carcinogenesis or other genomic diseases.8–16 Nucleotide excision repair (NER), base excision repair (BER), mismatch repair (MMR), homologous recombination (HR), non-homologous end joining (NHEJ), and trans-lesion repair were included in different types of DNA damages. However, NER pathway is the major DNA repair pathway that repairs most of the bulky DNA damage by ultraviolet (UV) radiation, chemicals, environmental toxins, and chemotherapeutic drugs. 17
The key step of DNA damage repair is the initial damage recognition. Chromosome aberrations, including chromosome breaks or rearrangements such as deletion, duplication, inversion, and translocation, are reflective of genomic instability. DNA double-strand break (DSB) is the most severe lesion to genomic DNA. Generally, both exogenous insults and endogenous insults can cause this kind of damage. If DSB failed to be repaired in time, genomic instability occurs to result in cell malignant transformation. 17
The function of a key NER protein, xeroderma pigmentosum group A–complementing protein (XPA), is to excise the damaged DNA and to repair it. 18 It has been found that XPA mutants are closely associated with the most severe clinical xeroderma pigmentosum symptoms. Once the cell recognizes DNA damage, a series of protein factors are produced, processed, and recruited to confirm the presence of damage, after which these proteins cleave the damaged nucleotide at 5′ and 3′ and fill in the gap to seal the resulting gap. 19 XPA is involved in these processes, and its recruitment to the damage site mediated by the transcription factor II H complex unwinds the double-stranded DNA at the site of the damaged nucleotide to form a NER bubble to facilitate repair. 20 In all these processes, XPA is believed to function for damage verification and for assembly of NER incision complexes. Moreover, XPA protein may be involved in the development of chromosome aberrations through DSBs repair pathway.
However, although XPA has been extensively studied for many years, 21 its involvement in the tumorigenesis of some cancers has been appreciated,22–25 and it is unknown whether XPA plays a role in the pathogenesis of BC. Here, we present evidence that XPA defect in DSB repair may result in increased chromosome aberrations in BC.
Materials and methods
Protocol approval
All experiments were carried out in strict accordance with the regulations in the guide for the experimental research issued by the Third Military Medical University. Ethical approval for the study was obtained from the Third Military Medical University in accordance with the Declaration of Helsinki (Ethical Principles for Medical Research Involving Human Subjects). Surgical specimens from 27 BC patients and matched adjacent NT were obtained in the Third Military Medical University from 2013 to 2015. All patients gave signed, informed consent for the specimens to be used for scientific research. The diagnoses of BC were based on both pathological and cytological evidence, evaluated by senior pathologists according to the World Health Organization classification criteria.
Cell line, transfection, and reagents
A human BC cell line RT4 was purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). RT4 was generated from a 65-year-old male Caucasian 26 and expressed modest levels of XPA. The RT4 cells were cultured in RPMI1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA) in a humidified chamber with 5% CO2 at 37°C. XPA, scrambled control (scr), and short hairpin small interfering RNA for XPA (shXPA) were all purchased from Origene (Beijing, China). The XPA or shXPA inserts were cloned into pSilencer 5.1-H1 Retro vector (Thermo Scientific, Rockford, IL, USA) using HindIII and EcoRI as restriction enzyme cutting sites, which were then used to transfect the cells at a concentration of 50 nmol/L using Lipofectamine 2000, according to the manufacturer’s instructions (Invitrogen). The transfection efficiency was more than 95%.
Reverse transcription quantitative polymerase chain reaction
Total RNA was extracted from the BC cells using RNeasy kit (Qiagen, Valencia, CA, USA). Complementary DNA (cDNA) was then randomly primed using 2 µg total RNA with an Omniscript reverse transcription kit (Qiagen). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was in duplicate using the QuantiTect SYBR Green PCR system (Qiagen). The primers for XPA (NM_000380, XM_006717278, QT00029519) and α-tubulin (NM_001172085, NM_003194, QT00000721) were purchased from Qiagen. The values of the genes were analyzed using 2-ΔΔCt method. The relative expression levels of genes were first normalized against α-tubulin and then compared to the experimental controls.
Western blot
The protein was extracted and homogenized in cold radioimmunoprecipitation assay (RIPA) lysis buffer (Invitrogen). The supernatants were then collected after centrifugation at 10,000g at 4°C for 10 min. Protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Bio-Rad, China), and whole lysates were mixed with 4× sodium dodecyl sulfate (SDS) loading buffer (Bio-Rad) at a ratio of 1:3, heated at 100°C for 5 min and were separated on SDS-polyacrylamide gels, transferred to a polyvinylidene difluoride (PVDF) membrane, blocked, and then probed sequentially with a primary antibody and a second antibody. Autoradiogram used the enhanced chemiluminescent system before signal detection. Primary antibodies for Western blot are anti-cyclin-dependent kinase 1 (CDK1), anti-cyclin-dependent kinase 2 (CDK2), anti-Cyclin B, anti-Cyclin E, and α-tubulin (all from Cell Signaling, San Jose, CA, USA). Secondary antibody is horseradish peroxidase (HRP)-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). Densitometry of Western blots was quantified using NIH ImageJ software (Bethesda, MA, USA). The protein levels were first normalized to α-tubulin and then normalized to experimental controls.
Cell counting kit-8 assay
A cell counting kit-8 (CCK-8) detection kit (Sigma-Aldrich) was used to analyze cell viability according to the manufacturer’s instructions.
The cytokinesis-block micronucleus test
The cytokinesis-block micronucleus test (CBMNT) assay was performed as described earlier. 27 Briefly, the cells were treated with chromosome medium A, supplemented with 2% phytohemagglutinin (PHA)-L at 37°C for 48 h, after which 6 µg/mL Cytochrome b (CytB) was added for 24 h before harvest and analysis. Cells were then fixed with methanol/glacial acetic acid/0.9% NaCl (5:1:6), air-dried, and then stained with 125 µg/mL acridine orange in phosphate-buffered saline (PBS). DNA damage events are scored on mononuleated (MN) cells, binucleated (BN) cells, micronuclei (MN), nucleoplasmic bridges (NPB), and nuclear buds (NBUD).
Immunohistochemistry, immunofluorescence, and gamma-H2AX foci detection
The bladder tissue from the patients was staining with Fast Red (Sigma-Aldrich). Cellular immunofluorescence was staining routinely. For gamma-H2AX foci detection for DNA DSB, cells were grown on poly-
Fluorescence in situ hybridization analysis of MN
The origin of MN was assessed by fluorescence in situ hybridization (FISH; Cambio, Cambridge, UK). The biotin-labeled probe was detected with fluorescein isothiocyanate (FITC)-avidin and the signals were enhanced by biotinylated anti-avidin and FITC-avidin. The cells were counterstained with DAPI/propidium iodide.
Comet assay
Comet assay was applied to measure cellular deoxyribonucleic acid strand breaks, using a Comet Assay Kit (Cell Biolabs, Inc., San Diego, CA, USA), according to the manufacturer’s instructions. Briefly, the cells were embedded in agarose on a microscope slide and then lysed to form nucleoids containing supercoiled loops of DNA linked to the nuclear matrix. Electrophoresis at high pH results in structures resembling comets, which was captured by fluorescence microscopy for evaluation of DNA breaks.
Statistics
All statistical analyses were carried out using the GraphPad Prism 6.0 statistical software package (GraphPad Software, Inc., La Jolla, CA, USA). All values are depicted as mean ± standard deviation (SD) and are considered significant if p < 0.05. All data were statistically analyzed using one-way analysis of variance (ANOVA) with a Bonferroni correction, followed by Fisher’s exact test for comparison of two groups.
Results
XPA levels significantly decrease in BC
Although the involvement of XPA in the tumorigenesis of some cancers has been appreciated, it is unknown whether XPA plays a role in the pathogenesis of BC. Here, we aimed to address this question. We analyzed the levels of XPA in 27 BC patients and matched adjacent NT, by immunohistochemistry and by RT-qPCR. We found that the XPA levels were lower in BC tissue compared to NT (Figure 1(a)). Moreover, the XPA messenger RNA (mRNA) significantly decreased in BC compared to NT (Figure 1(b) and (c)).

XPA levels significantly decrease in BC. (a) Representative images for immunohistochemistry for XPA in BC and matched adjacent non-tumor bladder tissues (NT). (b and c) The RT-qPCR for XPA mRNA in BC and NT, shown (b) by individual levels and (c) by mean and SD.
Chromosome aberrations are detected in BC
Since XPA controls DNA repair, we examined the chromosome aberrations in cells isolated from BC, compared to NT, using FISH. We found that BC cells contained significantly more chromosome aberrations (chromosome number 3, 7, 17, and 9p16), shown by quantification (Figure 2). Thus, the loss of XPA in BC occurs concomitantly with increases in chromosome aberrations.

Chromosome aberrations are detected in BC. FISH was used to examine the chromosome aberrations in cells isolated from BC and NT, shown by quantification.
Modulation of XPA levels in BC cell line
In order to study the role of XPA in BC, we either overexpressed XPA in a BC cell line RT4, or depleted XPA in RT4 cells, by transfecting cells with a transgene or a shRNA (Figure 3(a)). Next, we confirmed the alteration of XPA levels in RT4 cells, by RT-qPCR (Figure 3(b)), by Western blot (Figure 3(c)), and by cellular immunofluorescence (Figure 3(d)).

Modulation of XPA levels in BC cell line. (a) Restriction map of the plasmid backbone used to overexpress or deplete XPA. (b–d) The XPA levels in RT4 cells, (b) by RT-qPCR, (c) by Western blot, and (d) by cellular immunofluorescence.
Loss of XPA increases BC cell viability upon DNA damages
Next, we examined whether XPA levels may affect the BC cell viability upon DNA DSB. We used two reagents, bleomycin (at 5, 10, 20, 40, 80 µg/mL) and etoposide (at 0.6, 1.25, 2.5, 5, 10 µmol/L), at different doses, to treat XPA-modified RT4 cells. Bleomycin is a non-ribosomal peptide that is toxic to BC cells through induction of DNA strand breaks and prevention of incorporation of thymidine into DNA strands in BC cells. Etoposide forms a ternary complex with DNA and the topoisomerase II enzyme to prevent re-ligation of the DNA strands, resulting in breaks of DNA strands to break. We found that XPA reduced BC cell viability upon these insults, while XPA depletion increased BC cell viability in a CCK-8 assay (Figure 4(a) and (b)).
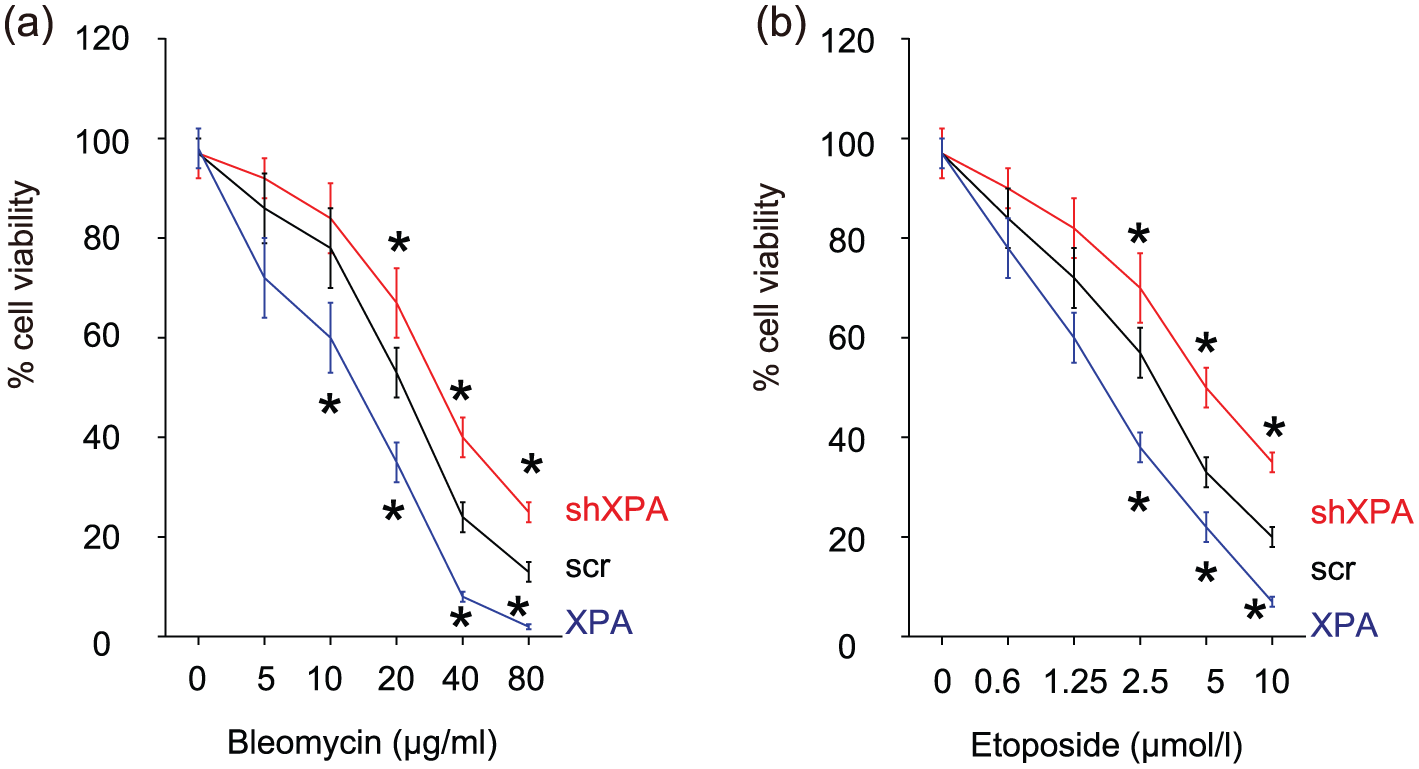
Loss of XPA increases BC cell viability upon DNA damages. (a) and (b) Two reagents, (a) bleomycin and (b) etoposide, at different doses, were used to induce DNA double-strand break in XPA-modified RT4 cells. The cell viability was quantified in a CCK-8 assay.
Loss of XPA impairs DNA repair
Then, we analyzed the effects of modulation of XPA on the BC cells in a CBMNT, a system for studying DNA damage, cytostasis, and cytotoxicity. Representative DNA damage events are once-divided BN cells (to compare with mononucleated cells); MN, a biomarker of chromosome breakage and chromosome loss; NPB, a biomarker of DNA false repair; and NBUD, a biomarker of elimination of DNA repair complexes (Figure 5(a)). We found that XPA decreased the events of MN (Figure 5(b)), NPB (Figure 5(c)), and NBUD (Figure 5(d)), while XPA depletion increased the events of MN (Figure 5(b)), NPB (Figure 5(c)), and NBUD (Figure 5(d)). These data suggest that loss of XPA impairs DNA repair.

Loss of XPA impairs DNA repair. DNA damages and repair were examined in a cytokinesis-block micronucleus test (CBMNT). (a) Representative DNA damage events are once-divided binucleated (BN) cells (to compare with mononucleated cells); micronuclei (MN), a biomarker of chromosome breakage and chromosome loss; nucleoplasmic bridges (NPB), a biomarker of DNA false repair; and nuclear buds (NBUD), a biomarker of elimination of DNA repair complexes. (b–d) Quantification of the events of (b) MN, (c) NPB, and (d) NBUD. Red arrows point to a specific damage.
XPA reduces formation of gamma-H2AX foci upon DNA DSB
DNA damage creates double-stranded breaks, followed by the phosphorylation of the histone, H2AX, which is a critical component of the histone octomer in nucleosomes. The phosphorylation of H2AX, which forms gamma-H2AX, is the first step in recruitment and localization of all DNA repair proteins. We thus examined the presence of gamma-H2AX in bleomycin-treated RT4 cells in a gamma-H2AX foci assay. We found that XPA decreased the foci formation by bleomycin, while XPA depletion increased it, shown by representative images (Figure 6(a)) and by quantification (Figure 6(b)).

XPA reduces formation of gamma-H2AX foci upon DNA double-strand break. (a and b) The presence of gamma-H2AX in bleomycin-treated RT4 cells was examined in a gamma-H2AX foci assay, shown (a) by representative images and (b) by quantification.
Changes in XPA levels affect DNA damages and repair in a comet assay
We then examined whether changes in XPA levels affect DNA damages and repair in a comet assay, also known as single cell gel electrophoresis assay, which is an uncomplicated and sensitive technique for the detection of DNA damage at the level of the individual eukaryotic cell. Shown by representative images (Figure 7(a)), we found that XPA significantly decreased the tail length (Figure 7(b)), tail integrity (Figure 7(c)), and tail distance (Figure 7(d)), while XPA depletion significantly increased the tail length (Figure 7(d)), tail integrity (Figure 7(c)), and tail distance (Figure 7(d)).

Changes in XPA levels affect DNA damages and repair in a comet assay. (a–d) DNA damages and repair were examined in a comet assay, shown by (a) representative images, (b) quantification for the tail length, (c) tail integrity, and (d) tail distance.
XPA may affect CDK1/Cyclin B and CDK2/Cyclin E to regulate cell cycle
Since the centrosome mutation results from both over-amplification of centriole and mis-regulation of the cell cycle, we hypothesize that XPA may affect G1 cell transition through CDK1/Cyclin E and may affect G2/M block through CDK2/Cyclin B. 28 We thus examined the changes in these cell-cycle regulators in XPA-modified RT4 cells. Shown by representative images (Figure 8), we found that XPA significantly decreased the levels of CDK1, CDK2, Cyclin E, and Cyclin B, while XPA depletion significantly increased the levels of CDK1, CDK2, Cyclin E, and Cyclin B.

XPA may affect CDK1/Cyclin B and CDK2/Cyclin E to regulate cell cycle.
Discussion
BC is the most common tumor of urinary system in the Chinese population, and its incidence is significantly higher than in Western countries. 9 BC is characterized with high recurrence rate, heterogeneity, and drug resistance. Chromosome aberrations are frequently found in BC cells, such as loss of heterogeneity, amplification, and aneuploidy, a common hallmark of human cancer. At present, DNA damage response and carcinogenesis have become a hot topic in tumor research field. Thus, identification of novel mechanisms underlying DNA damage repair in DNA damage response (DDR) is helpful for elucidation of carcinogenesis and high genomic instability.
XPA has a strong association with the formation of the NER bubble, in which XPA facilitates the assembly and structural organization of the NER incision complexes. Moreover, XPA actively interacts with other NER regulatory proteins and other DNA processing proteins. Mutations in XPA give rise to a defect in NER structure and function, resulting in severe problems.18–20 Furthermore, XPA also participates in the double-strand DNA repair.18–20
It has been increasingly recognized that the molecular mechanisms underlying the progressive resistance to treatment with DNA-damaging agents in cancer therapy may at least partially arise through the adaption of the DNA damage repair pathways, in which XPA has been found to play a key role. We assumed that XPA defect may change the capacity of DNA repair, which may trigger (pre)oncogenes and may inactivate tumor suppressor genes.
Here, we studied the role of XPA in the carcinogenesis of BC. Our first evidence came from the findings of the analysis on BC specimens from the patients. Decreased XPA was detected in BC, and this phenotype was closely associated with chromosomal aberrations. These findings were the basis of this study and inspired us to investigate the role of XPA in DNA damage and repair in BC cells, which is highly related to the tumorigenesis. Next, we successfully established XPA-overexpressing and XPA-depleted cells using plasmid-mediated transfection of a transgene or a shRNA. We confirmed the alteration of XPA levels in RT4 cells by RT-qPCR, Western blot, and cellular immunofluorescence.
Next, we tested BC cell viability upon DNA damages using two reagents, bleomycin and etoposide. We found that XPA reduced BC cell viability, while XPA depletion increased BC cell viability in a CCK-8 assay.
Then, we test several BC cell lines, including RT4, T24, and TCCSUP cell lines. We finally chose RT4 in our study, since XPA levels in RT4 is between T24 (high) and TCCSUP (low), and appeared to be appropriate for both gain-of-function and loss-of-function studies. The details of these pretests were not shown.
We performed a set of well-established techniques, including CBMNT, gamma-H2AX foci formation, and comet assay. In CBMNT, XPA decreased the events of MN, NPB, and NBUD, while XPA depletion increased the events of MN, NPB, and NBUD. The increased NPB in the loss of XPA suggested NHEJ repair pathways might be up-regulated, and error-prone repair pathway was regulated to respond to DSB. The increased NBUD in the loss of XPA suggested HR repair pathways might be downregulated, and the high-fidelity repair way was then modulated to repair DSB. Meanwhile, XPA decreased the foci formation by bleomycin, while XPA depletion increased it in the gamma-H2AX foci assay. Based on the findings in these experiments, we conclude that changes in XPA levels indeed affect DNA repair after damages, and loss of XPA reinforces the false repair to tumorigenesis.
Finally, we analyzed the relationship between XPA and cell-cycle regulators. The centrosome mutation represents a major mutation in bladder cells that give rise to cancer formation.29–32 Both over-amplification of centriole and mis-regulation of the cell-cycle-associated genes are responsible for the development of centrosome mutation. 9 CDK1/Cyclin E regulates the G1-S transition in the cell cycle, and CDK2/Cyclin B is associated with the G2/M phase shift. Both the processes are important for the development of centrosome mutation. 28 Since we detected that all these proteins were induced by loss of XPA, our data suggest that the effects of XPA on carcinogenesis in BC may be partially through its regulation on these cell-cycle regulators.
Together, our data suggest that high mutation frequency is the basis of mutations of oncogenes and anti-oncogenes and may be the essence of BC susceptibility. Thus, downregulated XPA may contribute to the carcinogenesis of BC via impairment of DNA repair.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by National Natural Science Foundation of China (No. 81302222) and Natural Science Foundation of Chongqing (cstc2013jcyjA10141).