
Abstract
Studies have demonstrated that microRNA 185 may be a promising therapeutic target in liver cancer. However, its role in hepatocellular carcinoma is largely unknown. In this study, the proliferation of human HepG2 cells was inhibited by transfection of microRNA 185 mimics. Cell-cycle analysis revealed arrest at the G0/G1 phase. Transfection of HepG2 cells with microRNA 185 mimics significantly induced apoptosis. These data confirmed microRNA 185 as a potent cancer suppressor. We demonstrated that microRNA 185 was a compelling inducer of autophagy, for the first time. When cell autophagy was inhibited by chloroquine or 3-methyladenine, microRNA 185 induced more cell apoptosis. MicroRNA 185 acted as a cancer suppressor by regulating AKT1 expression and phosphorylation. Dual-luciferase reporter assays indicated that microRNA 185 suppressed the expression of target genes including RHEB, RICTOR, and AKT1 by directly interacting with their 3′-untranslated regions. Binding site mutations eliminated microRNA 185 responsiveness. Our findings demonstrate a new role of microRNA 185 as a key regulator of hepatocellular carcinoma via autophagy by dysregulation of AKT1 pathway.
Introduction
Human hepatocellular carcinoma (HCC) is one of the few cancers with a continued increase in incidence. 1 HCC is a highly aggressive tumor with frequent distant metastasis and, therefore, is one of the world’s most dreadful diseases with poor prognosis. Patients are treated with powerful medications, using chemotherapy with side effects that are as challenging as the disease. 2 Surgery is another option. However, the mechanisms of HCC remain poorly investigated. Identification of novel diagnostic biomarkers and development of effective therapeutic interventions for HCC are urgently needed. The focus is on safe and accurate diagnosis and the development of treatment systems according to the unique complexities of the patient population. 3 Advances in nonsurgical treatment and better standardization with respect to diagnosis and patient eligibility for liver transplantation have been reported.4,5 For example, microRNAs (miRNAs) 6 may be novel biomarkers for HCC progression.
MiRNAs, which are approximately 22 nucleotides (nt) long, are incorporated into the RNA-induced silencing complex to induce translational repression or degradation of targeted messenger RNA (mRNAs). MiRNAs affect the expression of hundreds of targets and control gene expression patterns.7,8 Based on the recent research, several recent studies reported that miRNAs are stably detectable in plasma and serum.9,10 MicroRNA 185 (miR-185), which is located at 22q11.21, was described first as a regulator of cancer progression. For instance, overexpression of miR-185 suppressed the migration and invasiveness of LNCaP prostate cancer cells. 11 Accumulating evidence reveals the connection between miR-185 and cancers, including non-small cell lung cancer, 12 melanoma, 13 pediatric renal tumors, 14 breast cancer, 14 and glioma. 15 Liu et al. 16 reported that miR-185 acts as a growth suppressor in colorectal cancer cells by targeting RhoA and Cdc42 expression. Tang et al. 17 suggested that plasma miR-185 has become potential biomarker for glioma. Increasing evidence also suggests that miR-185 is essential for the regulation of liver development.3,4,6,18–21 However, the study is mainly focused on its role in lipid metabolism. Wang et al. 18 found that miR-185 plays an important role in regulating fatty acid metabolism and cholesterol homeostasis in hepatocytes, in addition to improving insulin sensitivity, both in vitro and in vivo. The previous study 22 showed that hepatitis C virus (HCV) core protein decreases the expression of miR-185-5p. And HCV core protein inhibits HepG2 cell apoptosis via the Sirt1–p53–bax pathway studied by Feng et al. 23 Therefore, we had the hypothesis that miR-185 may have relationship with HepG2 cell apoptosis. While Budhu et al. 21 identified a 20-miRNA tumor signature associated with HCC intravenous metastasis to predict HCC survival and recurrence in patients with multi-nodular or solitary tumors, miR-185 is one of the significantly differentially expressed miRNAs in human HCC tissues compared with non-neoplastic liver parenchyma.
Autophagy is an intracellular process, in which cells generate energy and metabolites by digesting their own organelles and macromolecules, to accommodate rapid cell growth and proliferation. Although apoptosis and autophagy are the two fundamental types of programmed cell death, the functional relationship between apoptosis and autophagy is complex. Under certain circumstances, autophagy constitutes a stress adaptation that avoids cell death (and suppresses apoptosis), whereas in other cellular settings, it constitutes an alternative cell-death pathway.
Until now, the potential link between miR-185 and autophagy remains unknown. We demonstrate here, for the first time, that miR-185 induces cell autophagy and apoptosis. When cell autophagy was inhibited by chloroquine (CQ) or 3-methyladenine (3-MA), miR-185 induced more cells apoptosis, suggesting miR-185 regulates cell autophagy to adapt circumstances and maintain its homeostasis. In this report, we identified miR-185-targeted oncogene AKT24,25 pathway in HCC, and further investigated its role in hepatic carcinogenesis. The results provide insight into the physiological and pathological mechanisms of miR-185 in HCC development.
Materials and methods
Cell culture
The hepatoblastoma cell line HepG2 was purchased from American Type Culture Collection (Manassas, VA, USA). All the cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS; Life Technologies, Grand Island, NY, USA), 100 U/mL of penicillin G, and 100 µg/mL of streptomycin (SV30010; Thermo Scientific, Rockford, IL, USA) at 37°C in a humid atmosphere of 5% CO2.
Target prediction
In order to improve the efficiency of prediction, conventional online programs, including Findtar (http://bio.sz.tsinghua.edu.cn) and Microrna (http://www.microrna.org/), were used to predict the targets of miR-185. The targets predicted by these two programs were further analyzed and demonstrated using the following experiments.
Plasmid construction, cloning, and mutagenesis of 3′-untranslated regions seed regions
The 3′-untranslated regions (3′-UTRs) of RHEB, RICTOR, and AKT1 were amplified by polymerase chain reaction (PCR) using genomic DNA of L02 cells as template and cloned into pmirGLO Dual-Luciferase miRNA Target Expression Vector (pmirGLO; Promega, E1330, Beijing 100013). The predicted target site was mutated by site-directed mutagenesis. Full-length 3′-UTRs of AKT1 (1106 bp), RHEB (1200 bp), and RICTOR (800 bp), containing the putative miR-185 binding site, were amplified by PCR using the following primers:
wt-AKT1 (sense): 5′-GAGCTCACAGATGACAGCATGGAGTG-3′;
wt-AKT1 (anti-sense): 5′-GCTAGCACTCTTCCACCCAGCAAAG-3′;
wt-RHEB (sense): 5′-GCTAGCAGCTTCACAAGGCAAGTCTT-3′;
wt-RHEB (anti-sense): 5′-CTCGAGCACAGAACACTAAACTCCAG-3′;
wt-RICTOR (sense): 5′-GCTAGCTTTCCCTAGATGCATGAAGA-3′;
wt-RICTOR (anti-sense): 5′-CTCGAGAAGAAAAACGTGAAAGGGC-3′.
The PCR product was subcloned into a SacI/NheI site of the pmirGLO basic luciferase reporter vector. A construct containing 3′-UTRs of AKT1, RHEB, and RICTOR with a mutant seed sequence of miR-185 was synthesized, respectively, using the following primers:
mut-AKT1 (sense): 5′-GAGCCTCCCC CTCAGATGATGTGTGCACGG TAGCACTTGA-3′;
mut-AKT1 (anti-sense): 5′-GTCGAAAAGG TCAAGTGCTACCGTGCACAC ATCATCTGAG-3′;
mut-RHEB (sense): 5′-ATTTGAGGGTGGAAGCACACAATTTCATCACT-3′;
mut-RHEB (anti-sense): 5′-AGTGATGAAATTGTGTGCTTCCACCCTCAAAT-3′;
mut-RICTOR (sense): 5′-TCAAAAGGCTCAACAAAGCACACAAGGAAGTTGTTTTGG-3′;
mut-RICTOR (anti-sense): 5′-CCAAAACAACTTCCTTGTGTGCTTTGTTGAGCCTTTTGA-3′.
MiRNA oligonucleotides
The chemically synthesized miR-185 mimics and negative mimic control were purchased from RiboBio Co., Ltd (Guangzhou, China).
Luciferase reporter assay
Cells were seeded in a 48-well culture plate, to about 80% confluence. Cells were transiently transfected with wild-type (wt) or mutant-type (mut) reporter plasmids or 100 mM miR-185 mimics using jetPRIMETM (Polyplus-transfection Inc., France) according to the manufacturer’s protocol. In addition, 25 ng of the Renilla luciferase vector (pRL-TK) DNA was transfected in each well. After 24 h of transfection, the cells were lysed in passive lysis buffer (E1941; Promega). Luciferase activities were measured in a microplate luminometer according to the technical manual for the Dual-Luciferase Reporter Assay System (E1910; Promega, USA).
RNA isolation, reverse transcription, and quantitative real-time-PCR
Total RNA from various transfected HepG2 cells was prepared using the Total RNA Kit (R6834; Omega, Norcross, GA, USA) according to the manufacturer’s instructions. Total RNA was reverse transcribed into single-stranded complementary DNA (cDNA) using the PrimeScript RT reagent Kit (DRR027A, TaKaRa, China). The cDNA was subjected to qPCR (4367659, ABI, USA) amplification using specific primers. The relative amounts of mRNAs were analyzed by quantitative PCR using SYBR Green qPCR Master Mix (1206352, Applied Biosystems, Warrington, UK) on an ABI 7500 System (Applied Biosystems, NY, USA). The optimized primers used for real-time PCR are listed in Table 1.
Primers used for real-time polymerase chain reaction.

Western blot
Cells were lysed in lysis buffer (Thermo, 78501, USA) containing 150 mmol/L NaCl, pH 7.5, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 50 mmol/L Tris–Cl, 10% glycerol, 1% NP-40, and a protease-inhibitor cocktail (5872s, CST) for 30 min on ice. Protein concentrations were determined using the Pierce BCA assay (23225, Thermo Scientific). Equal amounts of protein were separated by 12% Bis-Tris Gel/MOPS (NP0341, Invitrogen, Grand Island, NY, USA) or 10% sodium dodecyl sulfate (SDS)–polyacrylamide gels and electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane (ISEQ00010, Millipore, Billerica, MA, USA). After blocking with 5% nonfat dry milk (2321000, BD, USA) for about 3 h at room temperature or overnight at 4°C, the membranes were incubated with primary antibodies including anti-LC3B (3868, CST, USA), anti-beclin 1 (3495, CST, USA), anti-p-AKT1 (S473, D7F10, CST, USA), anti-PI3KIII (4362, CST, USA), anti-ATG3 (3415, CST, USA), anti-ATG5 (8540, CST, USA), anti-GAPDH (5174, CST, USA), anti-β-actin (sc-47778, Santa Cruz, USA), anti–mechanistic target of rapamycin (mTOR) (ab32028, Abcam), anti-p-mTOR (ab109268, Abcam) or anti-RHEB (ab92313, Abcam), and anti-RICTOR (53A2, CST, USA) in Tris-buffered saline containing 0.1% Tween-20. Membranes were washed three times with Tris-buffered saline (TBS)–Tween, and incubated with goat anti-rabbit (ZB-2301, 1:5000, ZSGB-BIO, China) or goat anti-mouse (ZB-2305, 1:5000, ZSGB-BIO, China) antibodies for 1.5 h at room temperature, and washed three times as before. Immunoreactive bands were detected using an enhanced chemiluminescence system (32209, Thermo Scientific). Western blot data were quantified using Image J software.
Proliferation assay
HepG2 cells were seeded at a density of 5000 cells/well (100 µL of DMEM with 10% FBS) on a 96-well plate, 24 h before transfection. Cell counting kit-8 (CCK-8) solution (EQ645; DOJINDO, Kumamoto, Japan) was added to each well at 24, 48, and 72 h, respectively, after transfection. The optical density was read at 450 nm according to the manufacturer’s instructions.
Flow cytometry
Cells were harvested at 48 h post-transfection with or without 3-MA or CQ added 6 h before harvested. Then cells were washed twice with cold BioLegend cell staining buffer (420201) before resuspension in Annexin V Binding Buffer (422201) at a concentration of 106 cells/mL. FITC Annexin V and 7-aminoactinomycin D (AAD; 420401) were added successively. DNA content was analyzed using 7-AAD. Flow cytometry was performed using a FACS Calibur (BD Biosciences, USA). Data analysis was performed using FlowJo Version 7.6.1.
Autophagy assays
Autophagy was visualized in HepG2 cells by cotransfection with pCMV6-AN-RFP-LC3 (pRFP-LC3) (RC100053; Origene, Rockville, MD, USA) and miR-185 mimics/mimic negative control. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; D9542; Sigma, St Louis, MO, USA), followed by confocal microscopy (LSM510Meta; Zeiss, Bergisch Gladbach, Germany). In most experiments, pRFP-LC3 punctate staining was assessed from at least six random high-power fields. A single random Z-section was used for each field, and at least 30 cells per sample were counted. Samples were analyzed in triplicate for each condition. An LC3 punctate was considered to be a totally isolated RFP-positive structure greater than 1 µm in diameter.
Statistical analysis
Statistical evaluation of the data was performed using paired Student’s t test. The data were expressed as mean ± standard error of the mean (SEM) of at least three independent trials. SPSS 17.0 software (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. A statistical difference of p < 0.05 was considered significant.
Results
MiR-185 suppresses HepG2 cell proliferation and attenuates cell-cycle progression
In order to determine the effect of miR-185 on cell growth, CCK-8 assay was performed, which allows sensitive colorimetric determination of the number of viable cells in cell proliferation and cytotoxicity. Transfection of HepG2 cells with miR-185 mimics significantly reduced cellular proliferation compared with control cells after transfection for 48 and 72 h, respectively (Figure 1(a)). The results showed that miR-185 mimics markedly suppressed the proliferation of HepG2 cells. Next, we examined the effect of miR-185 mimics on cell-cycle progression. HepG2 cells were synchronized at G1/S boundary by starvation for 12 h. By contrast, miR-185 mimics induced significant G1 phase arrest (Figure 1(b) and (c)). G1/S cell-cycle checkpoint represents a common step in the multistep progression of malignant tumors. Expression of cell-cycle-related proteins was analyzed by western blot. Cyclin D1 was increased, while cyclin A and cyclin B1 were significantly attenuated in cells transfected with miR-185 mimics (Figure 1(d)).

MiR-185 suppresses HepG2 cell proliferation and attenuates cell-cycle progression: (a) HepG2 cells were seeded into 96-well plates at a density of 3 × 103 cells per well, and overnight cells were transfected transiently with miR-185 mimics or negative mimic controls; after transfection for 24, 48, and 72 h, the effect of miR-185 mimics on HepG2 cell proliferation was evaluated by CCK-8 proliferation assays, respectively; (b) the control cells progressed normally into S and G2 phases, but the cells transfected with miR-185 mimics were arrested at G1 phase in HepG2 cells. Cell-cycle analysis was determined 48 h after transfection using 7-AAD staining flow cytometry; (c) numbers in each histogram indicated the percentage of cells in G0–G1, S, and G2–M cell-cycle phases; and (d) HepG2 cells were transfected with miR-185 mimics or a negative control (NC) for 48 h; cell lysates were analyzed by western blot, miR-185 mimics induced increase in Cyclin D1, but decrease in cyclin A and cyclin B1 protein levels (n = 3, *p < 0.05, **p < 0.01), and data represent mean ± SEM.
Apoptosis-promoting effects of miR-185 in HepG2 cells
Apoptotic cells exhibit Annexin V positivity. Therefore, the ratio of Annexin V–positive cells is used as an appropriate method to quantify apoptosis, which is determined by flow cytometry. Untreated HepG2 cells showed low rates of apoptosis. In contrast, compared with HepG2 cells transfected with control, the total Annexin V–positive HepG2 cells increased after transfection with miR-185 mimics (Figure 2(a)). Statistical analysis of the three experimental data showed increase in both early and late apoptosis of HepG2 cells after transfection for 48 h (p = 0.0011 and p = 0.0057), respectively (Figure 2(b)). Bax and bcl-2 mediate carcinogenesis of HCC. The ratio of bax and bcl-2 increase is a classic symbol of cellular apoptosis. HepG2 cells transfected with miR-185 mimics for 48 h showed significant downregulation in bcl-2 expression (Figure 2(c)). These results suggest that miR-185 induces apoptosis of HepG2 cells.

Apoptosis-promoting effects of miR-185 in HepG2 cells: (a) MiR-185 induces apoptosis. Cellular apoptosis was detected by Annexin V/7-AAD combined labeling flow cytometry in HepG2 cells 48 h after transient transfection, (b) apoptotic evaluation was determined by the percentage of apoptotic cell number in total cell number. Annexin V–positive human HepG2 cells represented apoptotic cells. The apoptosis rate dramatically increased compared to that for the mimic control group. The results shown are mean ± SEM values (n = 3, *p < 0.05, **p < 0.01), and (c) the expression of apoptosis-related proteins bcl-2 and bax after transfection with miR-185 mimics in HepG2 cells were measured by western blotting. Bcl-2 was significantly downregulated, while the expression of bax was not altered significantly, an increased ratio of bax to bcl-2 was evident.
Overexpression of miR-185 induces autophagy
Autophagy and miRNA are important regulators of cancer cell tumorigenesis. Impaired autophagy and significantly altered expression of oncogenic miR-185 are observed in HCC. However, the relationship between the two phenomena remains mysterious. We speculated that miR-185 may have an essential role in cellular autophagy, which was validated by a RFP-LC3 punctate formation assay using confocal microscopy and an LC3 conversion assay using western blot (Figure 3(a) and (b)). HepG2 cells were transiently co-transfected with miR-185 mimics and pRFP-LC3. Transient transfection of miR-185 mimics increased pRFP-LC3 punctate formation. Quantification of RFP-LC3 dots in the cells established that transfection of miR-185 mimics induces autophagosome accumulation in HepG2 cells. In addition, western blot revealed the conversion of endogenous LC3-I to LC3-II consistent with early speculation and RFP-LC3 punctate formation. The intracellular levels of SQSTM1/p62 determined whether autophagosome accumulation was related to autophagy induction or inhibition of downstream pathways. Western blot showed that transfection of miR-185 mimics resulted in approximately 40% reduction in SQSTM1 protein expression in HepG2 cells (Figure 3(a)). Several autophagy-related genes (PI3KIII, ATG5, ATG3, and beclin 1) were detected by western blot (Figure 3(c)). The protein level was upregulated post-transfection with miR-185 mimics, which was consistent with the conversion of LC3-I to LC3-II, an important marker of autophagy.

MiR-185 mimics induce autophagy: (a) HepG2 cells were transiently transfected with miR-185 mimics or controls. Cell lysates were analyzed for p62, LC3B expression by western blot. The quantitative ratios were shown as relative optical densities of the bands normalized to GAPDH expression. The results represent mean ± SEM values (n = 3,*p < 0.05), (b) HepG2 cells were co-transfected with miR-185 mimics and pRFP-LC3, or mimic control and pRFP-LC3, alternatively. Punctate localization of LC3 on autophagic vacuoles was clearly visible in cells transfected with miR-185 mimics but not the control. LC3 punctate dots were counted and presented as a bar graph. Scale bars: 5 µm (n = 3, *p < 0.05) and (c) western blot analysis of ATG5, ATG3, PI3KIII, beclin 1, and GAPDH protein in HepG2 cells transfected with miR-185 mimics, or negative control (NC). Protein ratios were calculated following Image J densitometric analysis. Similar results were obtained in three independent experiments.
MiR-185-mediated autophagy suppresses HepG2 cell apoptosis
The proposition that miR-185 is involved in cell apoptosis is not totally new. With the purpose of detecting whether miR-185-mediated cell autophagy has relationship with cell apoptosis, 3-MA and CQ were used to inhibit autophagy, respectively. The 3-MA is an agent that block class III phosphatidylinositol 3-kinase-dependent autophagosome formation, which is capable of suppressing starvation induced autophagy. 26 CQ is a well-known inhibitor of autophagic protein degradation through autophagosome acidification and subsequent proteolytic digestion. 27 The LC3-II expression increased at 6 h after CQ treatment as western blot results showed (Figure 4(a)). And the conversion of endogenous LC3-I to LC3-II was inhibited, in 3-MA-treated HepG2 cells (Figure 4(a)). Significantly, both inhibitors exhibited autophagy inhibition-independent effects, and miR-185 induced autophagy.

MiR-185-mediated autophagy suppresses HepG2 cell apoptosis: (a) MiR-185-mediated autophagy could be inhibited by 3-MA and CQ. HepG2 cells were incubated for 24 h and transfected with miR-185 mimics (M5) or mimic control (MC) for 48 h, in the presence or absence of autophagy inhibitors chloroquine (CQ; 10 µM) or 3-methyladenine (3-MA; 10 mM) for 6 h before harvesting. LC3B conversion were analyzed by immunoblotting, (b) in order to explain whether autophagy induced by miR-185 is a cause or a consequence of apoptosis, we conduct experiments with miR-185 mimics in combination with autophagy inhibitors (CQ and 3-MA), then we not only assess apoptosis with flow cytometry for Annexin V (e and f), but also detect the expression of bcl-2 when autophagy was inhibited by (b) CQ, (c) 3-MA, and (d) si-beclin 1, and the results suggested specific mechanism of miR-185 role on autophagy and apoptosis.
In order to explain whether autophagy induced by miR-185 is a cause or a consequence of apoptosis, we next investigated the contribution of miR-185-mediated autophagy to HepG2 cell apoptosis. Through the blockage of autophagy by 3-MA or CQ, two autophagy-blocking agent (Figure 4(b), (c), (e) and (f)), HepG2 cell apoptosis was increased, suggesting that autophagy inhibited cell apoptosis, which may plays a role in balancing the survival of tumor cell. To confirm these data, beclin 1–small interfering RNA (siRNA) treatment was used to abrogate autophagy, as shown in Figure 4(d). Silencing beclin 1 caused attenuation of autophagy in the HepG2 cells, and the expression of bcl-2 was decreased. Correspondingly, the cell apoptosis, after treatment with beclin 1-siRNA was similar to that of cells treated with 3-MA and CQ. Collectively, these data indicated that miR-185-mediated autophagy could suppress HepG2 cell apoptosis.
Experimental identification of miR-185 targets
We used FindTar, a prediction algorithm designed by Tsinghua University, to identify the potential targets of miR-185 in preventing the formation of malignant tumors. FindTar predicted that several regulators of proliferation and apoptosis signaling, including AKT1, CDC42, CDK6, RHEB, RICTOR, and IGF2A, are potential miR-185 targets. Furthermore, RhoA, SIX1, CCNE1, and RAB5A were also putative miR-185 targets. The quantitative real-time polymerase chain reaction (qRT-PCR) was performed to examine the mRNA levels of miR-185 putative targets. MiR-185 mimics but not mimic control significantly attenuated the mRNA levels of AKT1, RICTOR, and RHEB. Other putative miR-185 target genes, however, did not show significant changes in HepG2 cells (Figure 5(a)). Western blots showed that the cellular levels of AKT1, RICTOR, and RHEB were decreased in cells transfected with miR-185 mimics (Figure 5(b)).
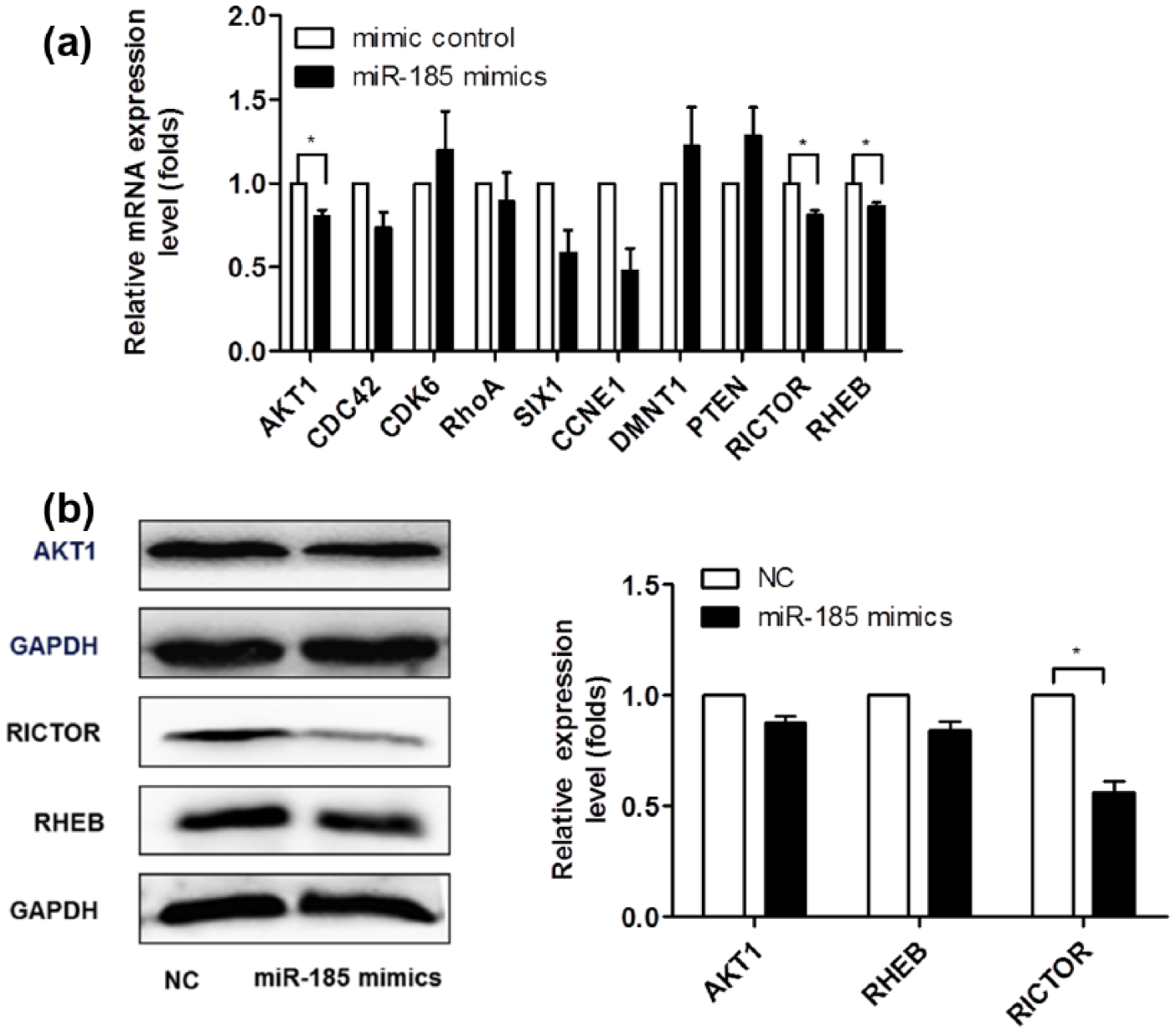
Experimental identification of miR-185 targets: (a) FindTar predicted that several regulators of the proliferation and apoptosis signaling pathway may be targets for miR-185. The qRT-PCR was performed to analyze the mRNA levels of miR-185 putative targets, and β-Actin was used as the internal control in real-time PCR. Only AKT1, RICTOR, and RHEB showed significant changes when transfected with miR-185 mimics in mRNA level and (b) AKT1, RICTOR, and RHEB protein levels were analyzed by western blot. The results are representative of three independent experiments and are presented as mean ± SEM values (n = 3,*p < 0.05, **p < 0.01). Western blot data were quantified using the Image J software.
MiR-185 directly interacts with 3′-UTRs of AKT1, RICTOR, and RHEB
The protein and mRNA levels of AKT1, RICTOR, and RHEB were downregulated in cells transfected with miR-185 mimics. Both TargetScan and FindTar predicted that miR-185 potentially binds to sequences in the 3′-UTRs of AKT1, RICTOR, and RHEB. In order to determine whether AKT1, RICTOR, and RHEB were miR-185 target genes, we constructed the pmirGLO-luciferase reporter containing either the wild-type (WT) or mutated (MUT) miR-185 binding sequences in the 3′-UTRs of the genes (Figure 6(a), (c), and (e)). Treatment with miR-185 mimics significantly reduced the activity of firefly luciferase with the wild-type but not mutant 3′-UTRs of AKT1, RICTOR, and RHEB (Figure 6(b), (d), and (f)). These results strongly indicate that AKT1, RICTOR, and RHEB are direct target genes of miR-185.

MiR-185 directly interacts with the 3′-UTRs of AKT1, RICTOR, and RHEB: (a, c, e) predicted binding sequences involving miR-185 and seeds matches in AKT1, RHEB, and RICTOR 3′-UTRs and (b, d, f) luciferase reporter vectors were generated by inserting the wild-type or mutant 3′-UTR fragments of AKT1, RHEB, and RICTOR into pmirGLO plasmid. Luciferase reporter assays at 24 h after transfection with wild-type (WT) or mutant (MUT) plasmids co-transfected with control or miR-185 mimics. Data shown represent means ± SEM of independent experiments, n = 3, **p < 0.01, *p < 0.05.
Role of miR-185 in AKT signaling
To determine the role of miR-185 in AKT signal transduction, we used a phospho-Ser473 antibody to directly measure the activation of AKT1. The phosphorylation of AKT1 proteins was significantly reduced when cells were transfected with miR-185 mimics, as well as the protein levels of mechanistic target of rapamycinmTOR and the phosphorylation of mTOR (Figure 7(b) and (c)). Overexpression of miR-185 did not alter the upstream expression of PTEN or phosphatidylinositol 3-kinase (PI3K) levels (Figure 7(a) and (c)).

Role of miR-185 in AKT signaling: (a and b) MiR-185 mimics or negative control (NC) were transiently transfected into HepG2 cells and cells were harvested at 48 h for western blot analysis. The upstream regulators such as PTEN and PI3K (a) and the downstream regulator mTOR, (b) of AKT signal pathway were measured and (c) protein ratios were calculated following Image J densitometric analysis, and the results from three independent experiments were similar.
MiR-185 is a potent cancer suppressor
MiR-185 acts as a formidable cancer suppressor by targeting multiple players in the AKT pathway, which control cellular functions, including cell proliferation, apoptosis, and progression of cell cycle and autophagy. The functional relationship between apoptosis and autophagy is complex. Under certain circumstances, autophagy constitutes a stress adaptation that avoids cell death and suppresses apoptosis, whereas in other cellular settings, it constitutes an alternative cell-death pathway. Our results showed when miR-185-mediated cell autophagy was inhibited, cell apoptosis would be promoted (Figure 8).
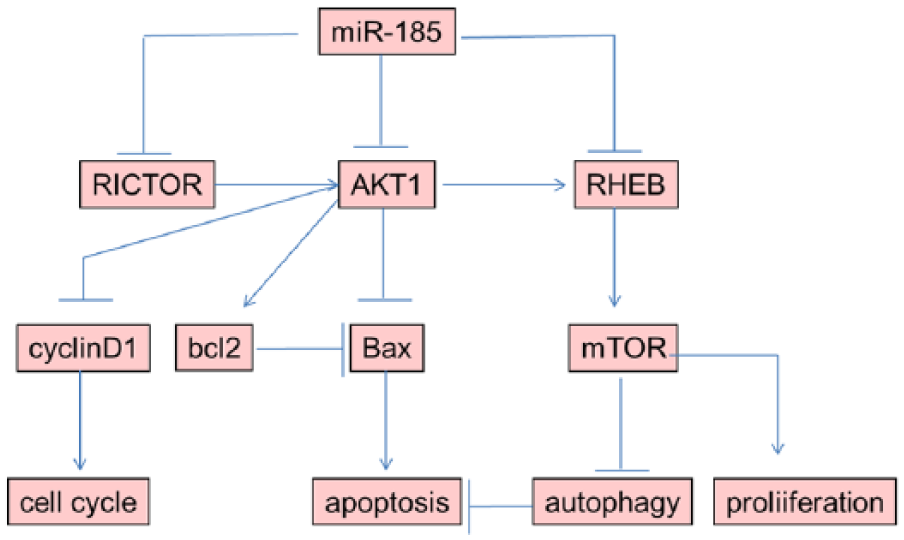
Schematic image.
MiR-185 targets several genes of AKT pathway to regulate cell proliferation, apoptosis, and cell-cycle progression and autophagy. When miR-185-mediated cell autophagy was suppressed, cell apoptosis would be promoted.
Discussion
A serine/threonine kinase known as AKT/protein kinase B has been identified as an important component of pro-survival signaling, which play key roles in cell proliferation, survival, and metabolism. In this study, we found that miR-185 plays an important role in the regulation of AKT pathway in HepG2 cells (Figure 8). Luciferase assay indicated that AKT1 is one of the miR-185 target genes (Figure 6(a) and (b)). AKT1 contributes to cell proliferation. Transfection with miR-185 mimics reduces cellular proliferation, and the expression of cyclin D1, a downstream gene of AKT1, was increased, resulting in cell-cycle arrest at G1 phase (Figure 1(b) and (c)). The levels of cyclin A and cyclin B1 were significantly reduced (Figure 1(d)). Blocking the cell cycle induces apoptosis. AKT1 is a major mediator of cell survival through direct induction of pro-survival member bcl-2. MiR-185 induced HepG2 apoptosis via AKT1–bcl-2 signaling pathway (Figures 2(c) and 8). Transfection with miR-185 mimics for 48 h significantly downregulated bcl-2 protein expression in HepG2 cells (Figure 2(c)). Consistent with several previous studies,28,29 miR-185 plays a predominant cancer suppressor role. This pathway is therefore an attractive target for the development of novel anticancer agents.
Tumor microenvironment plays a crucial role in tumor cell proliferation and progression. As the tumor grows, the stressful conditions always induce autophagy, 30 which controls intracellular homeostasis. 31 In this process, long-lived proteins and whole organelles are sequestered in double-membrane vesicles, called autophagosomes, and digested. Autophagy is a catabolic process in which cells digest their own components to provide energy for normal cellular function under unfavorable conditions.32,33 In particular, the AKT pathway is a crucial regulator of survival during cellular stress.34,35 In this study, miR-185 directly targeted AKT1, and the phosphorylation of AKT1 and mTOR was suppressed in cells transfected with miR-185 mimics. They are important upstream regulators of MTORC1 and MTORC2. We further discovered that multiple components of AKT signaling represent novel miR-185 targets, including RHEB and RICTOR (Figure 6(c)–(f)). By suppressing the activation of AKT pathway, miR-185 induces autophagy (Figure 8). Transfection of miR-185 mimics accelerated RFP-LC3 dot formation, LC3-I to LC3-II conversion, and SQSTM1 degradation (Figure 3(a) and (b)). A potential link between miR-185 and autophagy has yet to be reported. We demonstrated here, for the first time, that miR-185 is a potent autophagy inducer in a significant series of analyses. MiR-185 targets, RHEB and RICTOR, are essential upstream regulators of MTORC1. The activity of MTORC1 is controlled by RHEB, a GTP-binding protein.36,37 RICTOR is a scaffold protein that regulates the assembly and substrate binding of MTORC2. 38 Phosphorylation of AKT1 at Thr308 leads to partial activation of AKT1, and phosphorylation of AKT1 at Ser473 by mTORC2 stimulates full enzymatic activity. The mTOR–RICTOR complex phosphorylates Ser473 of AKT/PKB, which is essential for full AKT/PKB activation. Thus, miR-185 is a potent autophagy inducer, which suppresses RICTOR- and RHEB-mediated MTORC1-AKT activity (Figure 8). Autophagy always occurs under stressful conditions. As the tumor grows, it rapidly outgrows its blood supply, leaving tumor cells deprived of oxygen and nutrition. Studies are needed to test the strategic role of miR-185 in the maintenance of homeostasis following environmental stress.
Apoptosis and autophagy-associated cell death are two fundamental types of programmed cell death. 39 In addition to the complex relationship between autophagy and apoptosis, autophagy is described as an important regulator of physiological events. 40 In our study, through the blockage of autophagy by 3-MA or CQ two autophagy-blocking agent, we found that HepG2 cell apoptosis was increased. Autophagy-related effects of miR-185 provide valuable information about its role under different conditions. In response to tumor metabolic stress, autophagy controls tumor necrosis and inflammation. Hence, autophagy serves as a cancer cell survival mechanism in established tumors. Consequently, the combination of autophagy inhibition with cancer therapy is proposed as a novel approach to potentiate tumor cell death. 41 Further studies are required to explore the contribution of miR-185 to chemotherapy sensitivity.
Additional work is required to define the mechanisms of autophagy and apoptosis in the control of abnormal cellular metabolism during cell development. In summary, this study provides novel evidence suggesting that miR-185 is a tumor-suppressor miRNA that induces HCC cell autophagy by targeting AKT1, RHEB, and RICTOR. Whether miR-185 and AKT1 can be concomitantly targeted for effective anti-HCC therapy, it remains to be determined. Because the AKT pathway is activated in many cancers, it is easier to inhibit activation than to replace lost tumor-suppressor function.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by grants from the National Natural Science Foundation of China (no. 81470863) and the Medical Specialty Development Projects of Beijing Municipal Administration of Hospitals (ZYLX201402 and DFL2015170).