
Abstract
This study aimed to explore the effect of coptisine on non-small-cell lung cancer and its mechanism through various in vitro cellular models (A549). Results claimed significant inhibition of proliferation by coptisine against A549, H460, and H2170 cells with IC50 values of 18.09, 29.50, and 21.60 µM, respectively. Also, coptisine exhibited upregulation of pH2AX, cell cycle arrest at G2/M phase, and downregulation of the expression of cyclin B1, cdc2, and cdc25C and upregulation of p21 dose dependently. Furthermore, induction of apoptosis in A549 cells by coptisine was characterized by the activation of caspase 9, caspase 8, and caspase 3, and cleavage of poly adenosine diphosphate ribose polymerase. In addition, coptisine was found to increase reactive oxygen species generation, upregulate Bax/Bcl-2 ratio, disrupt mitochondrial membrane potential, and cause cytochrome c release into the cytosol. Besides, treatment with a reactive oxygen species inhibitor (N-acetyl cysteine) abrogated coptisine-induced growth inhibition, apoptosis, reactive oxygen species generation, and mitochondrial dysfunction. Thus, the mediation of reactive oxygen species in the apoptosis-induced effect of coptisine in A549 cells was corroborated. These findings have offered new insights into the effect and mechanisms of action of coptisine against non-small-cell lung cancer.
Introduction
Non-small-cell lung cancer (NSCLC) accounting for about 85% of all lung cancer cases needs immediate attention because of the uncontrollable deaths of NSCLC patients in the world. 1 Although combination of chemotherapy and surgery has been identified as an optimal treatment for patients with early stage disease, no effective single-drug therapy is currently available. Hence, discovering specific and effective chemotherapeutic agents for NSCLC is a desperate need.
Globally, research to understand the control and spread of lung cancer cells has been intensified for the development of newer targeted therapy. Recently, chemotherapy using bevacizumab, erlotinib, cetuximab, crizotinib, and so on, is being used to treat NSCLC. Drugs like ganetespib, custirsen, and dacomitinib are presently under study for clinical usage. Researchers are even attempting to test drugs like sorafenib and sunitinib that were already approved for the treatment of other types of cancer. 2 Alongside, discovering newer lead molecules targeting natural products against NSCLC has also been stimulated.3–5 In line with this, our new outcomes on the effect of a plant-derived molecule, coptisine (COP), and its mechanism of action in NSCLC A549 cells are presented in this article.
COP, broadly belonging to isoquinoline group of alkaloids, was first reported by Awe Walther 6 while processing Chelidonium majus tincture. Subsequently, the presence of COP as a major constituent in plants such as Corydalis ternate, 7 Fumaria indica, 8 Chelidonii herba, 9 Coptic japonica, 10 Corydalis adunca, 11 Corydalis yanhusuo, 12 Stylophorum lasiocarpum, 13 and Coptis chinensis (Chinese Goldthread)8,14 has been identified. Previous pharmacological reports have described the antibiotic, 15 antibacterial, 16 antiviral, 17 and cardiovascular protection 18 properties of COP. Also, COP is an active ingredient in Chinese gold thread formulations selectively preventing vascular smooth muscle cell proliferation with GI50 of 3.3 µM. 19 Furthermore, it has been revealed to effectively inhibit proliferation of hepatoma, leukemia cells, 20 NSCLC, and ovarian cancer cells.19,21 Berberine, the congener of COP, has been well explored for the prevention of carcinogenesis and its therapeutic-enhancing effect with other anticancer therapies. 21 However, the mechanism underneath the COP-induced cytotoxicity targeting A549 cells was not explored. Hence, this study was undertaken.
Materials and methods
Materials
Dulbecco’s modified Eagle medium (DMEM), RPMI-1640, fetal bovine serum (FBS), trypsin/ethylenediaminetetraacetic acid (EDTA), bovine serum albumin (BSA), and 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from HiMedia Laboratories (Mumbai, India). Cell cycle reagent, in situ caspase 3/caspase 7 detection kit, and polyvinylidene fluoride (PVDF) membranes were purchased from Millipore (Billerica, MA, USA). Bcl-2, Bax, cdc2, cdc25C, cytochrome c (cyt c), cyclin B1, and pH2AX (ser 139) monoclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Annexin V/propidium iodide (PI) staining kit was purchased from eBioscience, Inc. (San Diego, CA, USA). 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA), N-acetyl-L-cysteine (NAC), Bradford reagent, rhodamine 123, and dimethyl sulfoxide (DMSO; molecular biology grade; assay >99.9%) were purchased from Sigma–Aldrich (St Louis, MO, USA). Caspase-9, caspase-8, poly adenosine diphosphate (ADP) ribose polymerase (PARP), p21, β-actin antibody, and horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Western blot chemiluminescence reagent was purchased from Thermo Scientific (Arlington Heights, IL, USA).
Cell lines and culture
The A549 human lung adenocarcinoma, H460 human large cell lung carcinoma cells, H2170 human lung squamous cell carcinoma, HT-29 human colorectal human carcinoma, MDA-MB-231 human breast cancer, and human umbilical vein endothelial cells (HUVEC) were obtained from the National Centre for Cell Science (Pune, India). The A549 and HT-29 cells were maintained using DMEM. Cell lines MDA-MB-231, H460, and H2170 were maintained using RPMI-1640 containing 10% FBS and 1% penicillin–streptomycin solution at 37°C in humidified atmosphere with 5% CO2.
Isolation of COP
COP was isolated from the seeds of Fumaria indica following the procedure described by Pandey et al. 8 The identification of COP was carried out through ultraviolet (UV), infrared (IR), 1H- and 13C nuclear magnetic resonance (NMR) spectral analysis. 8 The compound was ascertained as COP from its identity, established by performing co-TLC (mobile phase: CHCl3:MeOH:H2O:CH3COOH (85:15:1:2); Rf 0.4) and mixed melting point studies with the authentic sample. Furthermore, the NMR spectrum revealed the purity of the sample to be >95%.
MTT assay
A 100-mM concentration of COP was prepared by dissolving in DMSO, and subsequent concentrations ranging between 100 and 0.1 µM were prepared by diluting with cell culture medium. The final DMSO concentration used was less than 0.1% in every treatment. MTT assay was performed to assess cell proliferation effect of COP. 22 Briefly, 2500 cells/well were seeded in 96-well plate containing DMEM medium supplemented with 10% FBS and 1% penicillin–streptomycin. A series of COP concentrations were added and incubated for 48 h in the presence or absence of 5-mM NAC. After 48 h of incubation, 15 µL of MTT (5 mg/mL) was added to each well and incubated at 37°C for 4 h. Then, the supernatant was removed and 150 µL of DMSO was added to each well to dissolve the crystals. The absorbance was measured at 595 nm by Spectramax M4 plate reader (Molecular Devices, CA 94089, USA).
Cell cycle analysis
Flow cytometry following PI staining was used to measure the DNA content of treated cells. 23 A549 Cells were seeded in 6-well plates with 2.5 × 105 cells/well in DMEM supplemented with 10% FBS. After 18 h, cells were treated with either DMSO or various concentrations of COP (50, 25, and 12.5 µM) for 48 h. Cells were detached with trypsin EDTA and washed with ice cold PBS, fixed in 70% ethanol, and kept at −20°C till analysis. The cells were stained with cell cycle reagent containing PI according to manufacturer’s instructions. The fluorescence intensity of cells was analyzed by flow cytometer (Guava easyCyt; Millipore) (PI at Ex = 493 nm and Em = 636 nm).
Annexin V assay
To detect apoptosis in A549 cells after exposure to different concentrations of COP, annexin V/PI staining kit was used to distinguish live cells from early apoptotic cells (annexin V stained) and late apoptotic cells (stained with annexin V and PI). 24 Briefly, 0.25 million cells were seeded in 6-well plates and incubated with different concentrations (50, 25, and 12.5 µM) of COP for 48 h in the presence or absence of NAC. Cells were trypsinized and washed twice with ice cold PBS and resuspended in 200 µL of binding buffer. Then, 5 µL of annexin V and 5 µL of PI were added to each sample and incubated for 15 min in the dark. Finally, the fluorescence intensity of cells was analyzed using flow cytometer (Guava easyCyt; Millipore) (annexin V fluorescein isothiocyanate (FITC) at Ex = 485 nm, Em = 535 nm).
Caspase 3/caspase 7 estimation
Using CHEMICON® CaspaTag™ in situ caspase detection kit, caspase 3/caspase 7 activity in the cells was measured. This methodology was based on fluorochrome inhibitors of caspases (FLICA). 25 A549 Cells were plated at 0.25 million/well concentration in 6-well plates and treated with different concentrations of COP (50, 25, and 12.5 µM) for 48 h in the presence or absence of NAC. Following which the cells were incubated with FLICA reagent for 30 min. After incubation, cells were washed with PBS to remove excess reagent from cells and suspended them in PBS. Then, the intensity of fluorescence was measured at 485 and 520 nm emission on Perkin Elmer vector X3 plate reader (PerkinElmer Inc., MA 02451, USA).
Measurement of mitochondrial membrane potential
Rhodamine 123, a cationic dye, was used to detect changes in the mitochondrial membrane potential by flow cytometry. 26 Briefly, 1 × 106 cells/well were plated in 6-well plates for 24 h, following treatment with different concentrations (50, 25, and 12.5 µM) of COP in the presence or absence of NAC. Then, the cells were incubated with 2 µM of rhodamine 123 at 37°C for 30 min. Subsequently, the cells were harvested, washed, resuspended in PBS, and the fluorescence intensity of rhodamine 123 was measured using flow cytometer (Flowsight, Amnis, EMD Millipore).
Measurement of reactive oxygen species generation
To quantify the intracellular reactive oxygen species (ROS) levels, DCFDA, a non-fluorescent dye, was used. DCFDA turns to highly fluorescent 2′,7′-dichlorofluorescein upon oxidation by the ROS generated in the cell. 27 Briefly, 1 × 106 cells/well plated in 6-well plates were incubated with different concentrations of COP from 0.5 to 24 h. To test the effect of NAC on COP-triggered ROS, cells were pretreated with NAC for 1 h, and then the A549 cells were treated with 50-µM COP for 24 h. A volume of 10 µM of DCFDA was added 30 min prior to harvesting and then washed with PBS. Finally, the cellular uptake of ROS was measured using flow cytometer (Flowsight, Amnis, EMD Millipore).
Preparation of cytosolic fraction and mitochondrial fraction
Cells were grown in the presence of COP (50, 25, and 12.5 µM) or no drug in 6-well plates for 48 h. After incubation, the cells were washed with ice cold PBS, lysed in ice cold buffer (10-mM Tris-HCl; (pH 7.8), 1% nonidet P-40, 10-mM β-mercaptoethanol, 0.5-mM phenyl methyl sulfonyl fluoride, and cocktail protease inhibitor), and homogenized on ice. The homogenate was centrifuged at 1000 g for 10 min at 4°C. The supernatants were then centrifuged at 12,000 g for 30 min at 4°C and the resulting supernatants were finally collected as cytosolic fraction. The pellet was lysed in lysis buffer containing 10-mM Tris (pH 7.4), 150-mM NaCl, 1% Triton X-100, 5-mM EDTA (pH 8.0), and cocktail protease inhibitor. After centrifugation at 12,000 g for 30 min at 4°C, the supernatants were collected as mitochondrial fraction.
Protein extraction and western blot analysis
Cells (0.2 × 106 per well) were incubated with different concentrations of COP (50, 25, and 12.5 µM) for 48 h and were trypsinized and washed with ice cold PBS. The cell pellets were lysed in lysis buffer containing 50-mM Tris-HCl (pH 7.5), 150-mM NaCl, 0.5% sodium deoxycholate, 0.1% Triton X, 0.1% SDS, 25-mM sodium fluoride, 200-µM sodium orthovanadate, and 1X protease inhibitor cocktail for 30 min on ice. Cell lysates were sonicated and centrifuged at 14,000 g for 10 min at 4°C. Then, the total protein concentration of supernatants was estimated using Bradford protein assay kit. Equal amounts of protein (30–50 µg) heated in SDS sample buffer containing DTT at 100°C were loaded onto SDS-polyacrylamide gel and proteins electrophoretically transferred onto PVDF membrane. Membranes were blocked by incubation for 1 h with Tris-buffered saline (TBS-T; 25-mM Tris-HCl (pH 7.6), 150-mM NaCl, and 0.05% Tween 20) containing 5% BSA. Membranes were incubated with antibody at 4°C overnight in TBS-T containing 5% BSA followed by the corresponding HRP-linked secondary antibody at room temperature for 1 h in TBS-T containing 5% nonfat milk powder. Chemiluminescence substrate was then added to the membranes and the band intensity was calculated using Image J 1.42 (NIH, Bethesda, MD 20892, USA).
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM) of at least three independent experiments and statistically analyzed by two-way analysis of variance (ANOVA) followed by Bonferroni post test or t-test using the GraphPad Prism 5 software. The significance level was based on probability of p < 0.05, p < 0.01, and p < 0.001.
Results
Inhibition of cell proliferation by COP
The effect of COP on the proliferation of different human lung cancer cell lines including A549, H460, and H2170 was determined through MTT assay by exposing the cells for 48 h. Treatment with various concentrations of COP resulted in a dose-dependent inhibition of cell growth (Figure 1). COP exhibited IC50 values of 18.09, 29.50, and 21.60 µM against A549, H460, and H2170 cells, respectively. Furthermore, antiproliferative effect of COP against A549 cells was almost reduced when the cells were pretreated with a ROS scavenger NAC as compared to the corresponding groups which were not exposed to NAC. In order to test the cell specificity, the effect of COP against MDA-MB-231 and HT-29 cells was also tested, which demonstrated IC50 values of 20.15 and 26.60 µM, respectively. Also, a selectivity index of 7.2 was observed between the inhibitory effect against HUVEC and A549 cells. Thus, the study validated the antiproliferation effect of COP by testing on three different cancer cell lines and normal cell line.

COP-induced cytotoxicity in A549 cells. The cells were treated with indicated concentrations of COP for 48 h, with or without NAC addition, and the percentage of cell viability was estimated by MTT assay.
COP-induced DNA damage in A549 cells
To determine the effect of COP on the expression of pH2AX, a marker for DNA damage, pH2AX protein levels were estimated in A549 cells treated with different concentrations of COP using western blot assay. Results demonstrated a significant upregulation in the expression of pH2AX in a concentration-dependent manner in comparison with control (Figure 2).

Effect of COP on expression of pH2AX in A549 cells. Expression of pH2AX in A549 cells treated with indicated concentration of COP for 48 h was detected by western blot analysis and followed by densitometry. β-actin was used for loading control.
G2/M arrest in A549 cells by COP
The cytotoxic effect of COP due to cell cycle arrest was ascertained by performing flow cytometry. Cells were treated with 50, 25, and 12.5 µM concentrations of COP for 48 h which exhibited a concentration-dependent arrest of cells in G2/M phase of cell cycle. COP increased the cell population in G2/M phase to 36.8%, 29.9%, and 26.5% at concentrations of 50, 25, and 12.5 µM, respectively, compared with DMSO control (16.95%) (Figure 3(a)). This was accompanied by a decrease in cells in G1 phase observed at all concentrations. However, COP did not affect cells in S phase.

COP-induced G2/M arrest. (a) A549 cells were exposed to different concentrations of COP for 48 h stained with PI and analyzed by flow cytometry. (b) Expression of G2/M regulatory proteins, cyclin B1, cdc2, and cdc25C were detected by western blotting analysis followed by densitometry.
Effect of COP on G2/M regulatory proteins
To investigate the molecular mechanism of COP-mediated G2/M phase arrest, G2/M regulatory proteins such as cyclin B1, cdc2, cdc25C, and p21 in A549 cells were examined. Protein extracts were prepared from cells treated with different concentrations of COP for 48 h and analyzed by western blot assay. As a result, expression of cyclin B1, cdc2, and cdc25C were significantly reduced and p21 expression was significantly upregulated by COP in a dose-dependent fashion in comparison with control (Figure 3(b)).
Apoptosis by COP in A549 cells
To further decipher COP-induced cytotoxicity, FITC-conjugated annexin V and PI double staining were performed on both untreated and COP-treated A549 cells. In the untreated cells, annexin-labeled population (early apoptotic) was found to be 8.6% and both PI- and annexin-labeled population (late apoptotic) was observed to be 4.6%. While the early apoptotic population of cells was significantly increased to 58.5%, it was found to be 24.2% of cells in late apoptotic phase under higher concentration of COP, that is, 50 µM. Similarly, at 25 and 12.5 µM concentrations, 26.4% and 10.8% of cells were found to be early apoptotic, respectively (Figure 4(a) and (b)). Pretreatment with NAC significantly abrogated apoptotic effect of COP.

COP-induced apoptosis in A549 cells. (a) A549 cells treated with various concentrations of COP in the presence or absence of 5-mM NAC for 48 h. Then, the cells were double stained with FITC-conjugated annexin V and PI for flow cytometric analysis. (b) Quantitative data of (a).
Effect of COP on ROS generation
To investigate the role of ROS in COP-induced apoptosis, cells treated with COP were stained with DCFDH, and ROS was quantified by flow cytometry. The levels of ROS after COP treatment with 50 µM at 0.5, 1, 2, 4, 12, and 24 h were estimated to be 23.7%, 28.1%, 76%, 85.7%, 86.2%, and 87.6%, respectively. Results demonstrated the significant increase in the ROS levels in a time and dose-dependent manner by COP (Figure 5(a) and (b)). Further NAC significantly inhibited the COP-induced ROS generation (Figure 5(c)).

Effect of COP on ROS in A549 cells. (a) Cells were treated with different concentrations of COP from 0.5 h to 24 h and incubated with DCFDA for 30 min. The intracellular ROS was measured by flow cytometry. (b) Histogram depicting the percentage of ROS positive cells of indicated concentrations of COP at different time points. (c) Cells were pretreated with NAC for 1 h and then treated with COP (50 µM) for 24 h.
COP-induced mitochondrial dysfunction study
To determine the effect on mitochondrial function, change of mitochondrial membrane potential (MMP) by flow cytometer with rhodamine 123 was performed. Expressions of Bcl-2, Bax and cytochrome c were measured by western blot assay. Percentage of cells with MMP loss after COP treatment with 50, 25, and 12.5 µM at 24 h was found to be 71.2%, 36.5%, and 8.7%, respectively (Figure 6(a)). Additionally COP significantly increased the expression of cytochrome c in cytosol and reduced its expression in mitochondria along with the increase in Bax/Bcl-2 ratio in a dose-dependent manner (Figure 6(b)). The cells pretreated with NAC significantly revoked the disruption of mitochondrial membrane potential.
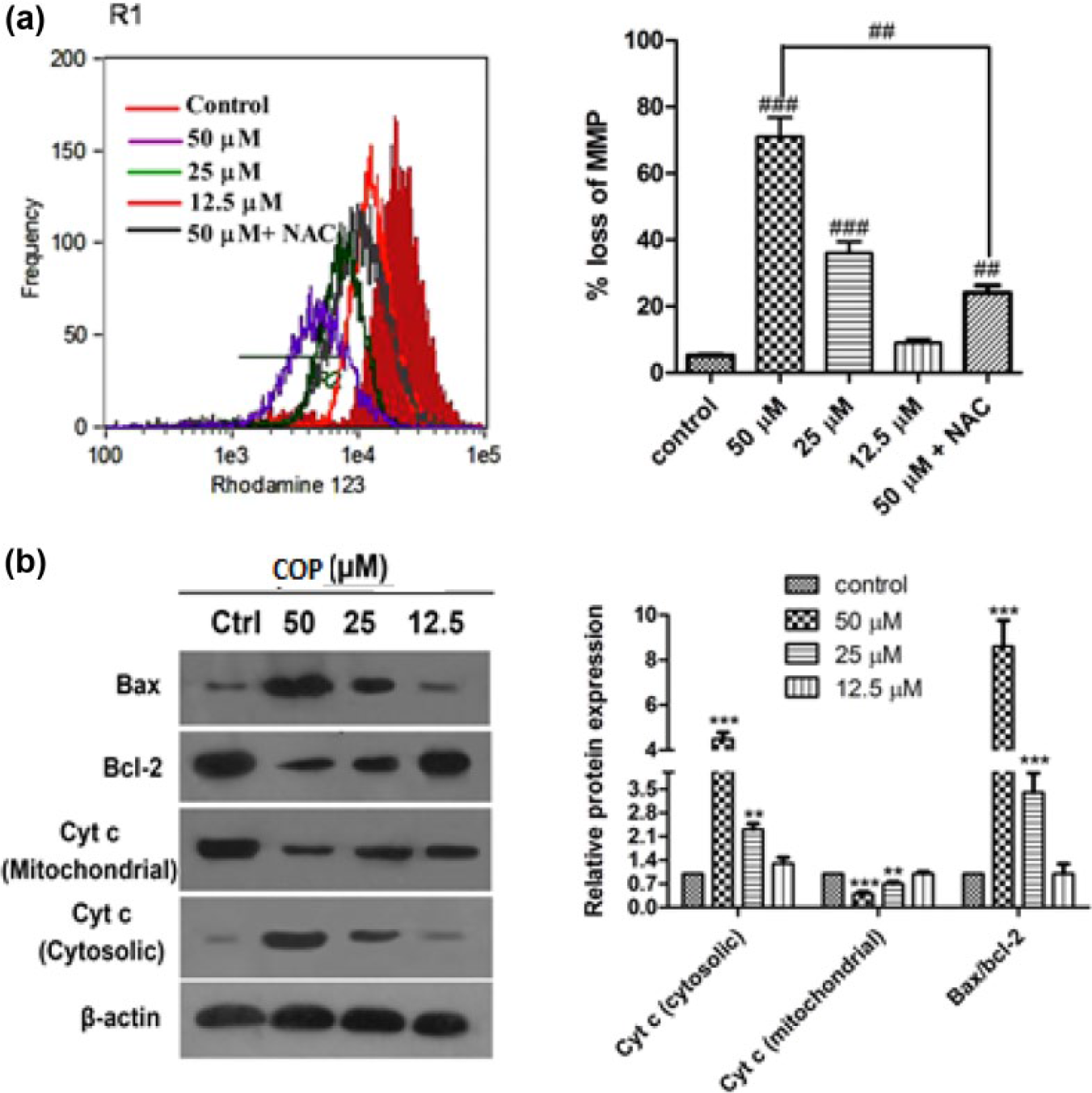
Effect of COP on mitochondrial function in A549 cells. (a) Cells were treated with different concentrations of COP with or without NAC for 24 h and then incubated with rhodamine 123 for 30 min. MMP was measured by flow cytometry. The histogram depicts COP-caused MMP loss in A549 cells and pretreatment with NAC prevented the COP-induced mitochondrial dysfunction. (b) Expression of Bax, Bcl-2, and cyt c in A549 cells were estimated by western blot and densitometric analysis. β-actin was used for loading control.
Effect of COP on activation of caspases
The mechanism of COP-induced apoptosis was further understood by examining the levels of caspase 3 through fluorescence assay, and the expression of caspase 8, caspase 9, and PARP by western blot assay. COP treatment increased the expression of active caspase 8 and caspase 9 along with the cleaved PARP dose dependently (Figure 7(b) and (c)). Active caspase 3 was increased by about 19-fold in cells treated with COP at 50-µM concentration compared to control, and dose-dependent induction of caspase 3 was observed. Induction of caspase 3 by COP was inhibited by NAC (Figure 7(a)).
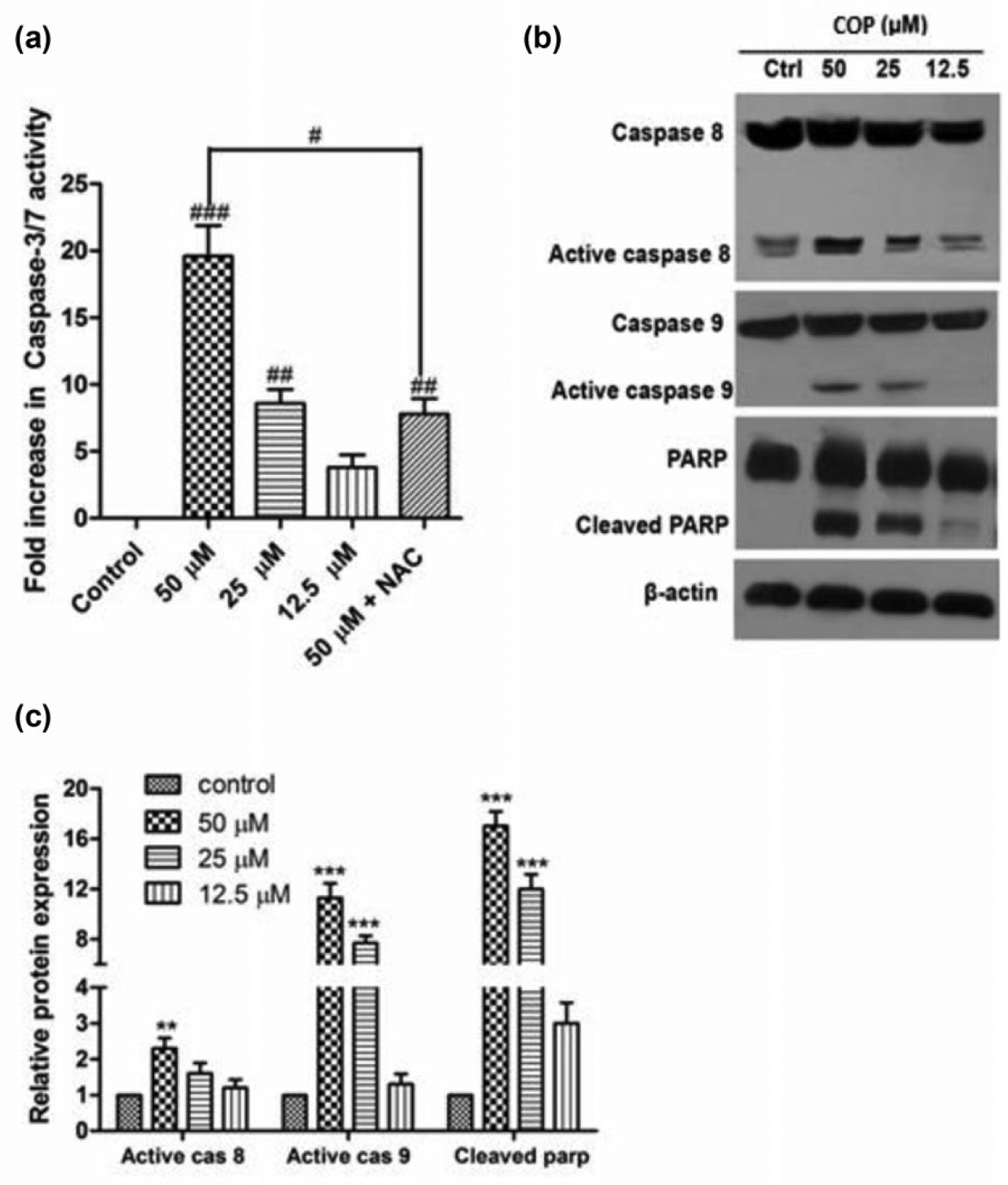
Effect of COP on activity of caspases. (a) Activity of caspase 3/caspase 7 was determined separately by in situ caspase assay kit. A549 cells were treated with different concentrations of COP in the presence or absence of 5-mM NAC for 48 h and fluorescence intensity was measured. (b) Expression of caspase 8, caspase 9, and PARP in A549 cells treated with indicated concentration of COP for 48 h was detected by western blot analysis c followed by densitometry. β-actin was used for loading control.
Discussion
Several plant-derived compounds have been recognized to possess anticancer activities both in vitro and in vivo. 28 Quercetin, a bioactive flavonol, was explored to inhibit cell proliferation and induce cell cycle arrest and apoptosis in different cancer cell types. In 2009, Chien et al. 29 investigated the human breast cancer MDA-MB-231 cell death mechanism of quercetin and reported it to be through mitochondrial- and caspase-3-dependent pathways. Deoxyelephantopin, a sesquiterpene lactone, had been spotted to exhibit antiproliferative and apoptosis-induced properties in SiHa cells. Recently, Farha et al. 30 had elucidated the underlying molecular mechanisms evidencing that STAT3/p53/p21 signaling, MAPK pathway, PI3k/Akt/mTOR pathway, caspase cascades, and ROS play critical roles in deoxyelephantopin-induced G2/M phase arrest and apoptosis of SiHa cells. Further compounds like artemisinin, sanguinarine, and cepharanthine have been proved to induce apoptosis through ROS-mediated mechanisms.27,31,32 Literature also claims hundreds of natural molecules screened for antiproliferative effects. However, only few among the several known cytotoxic natural compounds have been exploited for their mechanisms of action. Many potential molecules may emerge as chemotherapeutic drugs if relentless research is carried out on such underexploited natural compounds.
In view of this, COP, which was found to be cytotoxic against A549 cells, 20 was selected for exploring the mechanistic study. The procedures for the isolation and bulk separation of COP from various plant sources have been previously well explored.7,14 Also, COP possessing a quaternary iminium and tetraoxygenated protoberberine system has been reported to be possibly synthesized from its congener, Berberine. 8 The potentiality of COP in the treatment of cancer can be well recognized from its antiproliferative activity on hepatoma cell lines (HepG2, Hep3B, and SK-Hep1), leukemia (k562, u937, P3HI, and Raji), 19 NSCLC (A549), ovarian cancer (SK-OV-3), melanoma (HCT-15),20,21 and suppression of human breast cancer cell metastasis. 33 However, no mechanistic studies are delineated in the literature against A549 cells.
As a preliminary step, safety of COP was analyzed by performing cell viability test against HUVEC cells. Selectivity index (SI) value for COP was found to be 7.2, demonstrating its moderate safety. Also, the study involved testing of antiproliferative effect of COP against different cancer cell lines including A549, H460, H2170, MDA-MB-231, and HT-29. COP was found to be highly inhibitory toward A549 cells, compared to other tested cancer cell lines. Due to its more potency toward A549 cells, further investigation was performed on this particular cell line.
Chemotherapeutics were found to cause irreparable DNA damage which in turn induces cell death by activating checkpoint pathways and apoptotic pathways.34–37 Cell exposure to DNA damaging chemotherapeutic agents triggers the generation of double strand breaks which further results in phosphorylation of H2AX. Phosphorylation of H2AX at ser 139 is rapid and corresponds well with double strand break. Therefore, pH2AX is used as a marker to assess the DNA damage caused by chemotherapeutic agents. 38 To find whether COP can induce DNA damage, levels of pH2AX in treated A549 cells were estimated. Results revealed an increase in the expression of pH2AX, suggesting an induction of DNA damage by COP in A549 cells.
To elucidate the mechanism of antiproliferative activity of COP, the effects on cell cycle, as well as its ability to induce apoptosis, were evaluated. Cell cycle is controlled by numerous mechanisms ensuring correct cell division in normal cells while fundamental alterations in genetic control of cell division results in uncontrolled cell proliferation and eventually leading to the development of cancer. 39 The G2/M checkpoint prevents the cell from entering mitosis when DNA is damaged, providing an opportunity for repair and terminating proliferation of damaged cells. 40 Cyclins- and cyclin-dependent kinases (CDKs) are necessary for the cells to progress through the different phases of cell cycle. Cyclin B1 forms a complex with cdc2 (cdk1) that is essential for the cells to enter into mitosis. During G2phase, dephosphorylation of regulatory residues of cdc2, Thr14, and Tyr15, by phosphorylating cdc25C to pcdc25C at Ser216, directly activates cyclin B1/cdc2 complex and causes mitosis initiation. When the cells undergo genotoxic stress, phosphorylation of cdc2 weakens the activity of cyclin B1 and cdc2 complex that arrest cells in G2/M phase. 41 P21, a cyclin-dependent kinase inhibitor, can inhibit the growth of cancer cells by modulating the number of CDKs. It causes cell cycle arrest at G1/S or G2/M. Association of p21 with activated form of cdc2 results in reduction in cdc2 activity, suggesting its role in G2 arrest. 42 It was reported that COP does dual blockade of cell progression in vascular smooth muscle at G0/G1 and G2/M phases. 43 Effect of COP was further investigated on cell cycle distribution. Data revealed that COP caused cells to arrest only at G2/M in A549 cells in dose-dependent manner, suggesting that the effect of COP on cell cycle progression was cell type dependent. To further bolster our inference, the effect of COP was evaluated on expression of G2 regulatory proteins such as cyclin B1, cdc2, cdc25C, and P21. Significant reduction in the expression of cyclin B1, cdc2, and cdc25C, and upregulation of p21 confirmed that A549 cells were arrested at G2/M phase by COP.
In addition to cell cycle arrest, triggering of apoptosis by COP was found to be another important mechanism in inhibiting cell proliferation. Apoptosis serves as a protective mechanism that prevents the process of carcinogenesis resulted from mutations of genetic materials of normal cells. 44 Berberine, a related compound to COP, manifested inhibition of cell proliferation through apoptosis in different cell lines.21,45–47 Flow cytometric analysis delineated COP-induced apoptosis as a dose-dependent process.
ROS functions as redox messengers in intracellular signaling at physiological levels, while the excessive ROS generation can induce oxidative stress, loss of cell functioning, and apoptosis. 48 Discussions on the killing of cancer cells through ROS metabolism by increasing ROS levels above the toxic threshold are more particularly discussed in the literature.49–51 Excessive ROS accelerates mitochondrial depolarization and dysfunction, and it subsequently results in mitochondria-mediated apoptosis. 52 In the intrinsic pathway, mitochondria play a central role in activating apoptosis. While pro apoptotic members Bax or Bak form the mitochondrial apoptosis-induced channel (MAC), an early marker of onset of apoptosis, its formation is prevented by anti-apoptotic member Bcl-2. Formation of MAC on mitochondrial membrane causes release of cytochrome c to cytosol which triggers the activation of caspase 3 through activation of caspase 9. Caspase 3 is an activated death protease which cleaves PARP.53,54 In order to understand the mechanisms associated with the development of apoptosis, the levels of ROS, changes in mitochondrial function by detecting MMP, expression of Bcl-2 and Bax, and caspase cascades were estimated.
Present findings have suggested that the induction of ROS is an early event in the activity of COP in A549 cells. An increase in ROS production immediately 30 min post COP incubation was observed, and the induction of ROS was found to be increased dose and time dependent. Furthermore, COP-triggered ROS generation was significantly inhibited by the addition of NAC, which is generally used to identify and test ROS inducers. Moreover, COP significantly increased the ratio of Bax/Bcl-2, leading to the loss of MMP along with the release of cytochrome c into cytosol and activated caspase 9, caspase 3, and cleaved the PARP. Moreover, COP increased the expression of caspase 8 involved in extrinsic apoptotic pathway. These results clearly corroborated that mitochondrial apoptotic pathway is the mechanism of COP-mediated apoptosis (Figure 8).
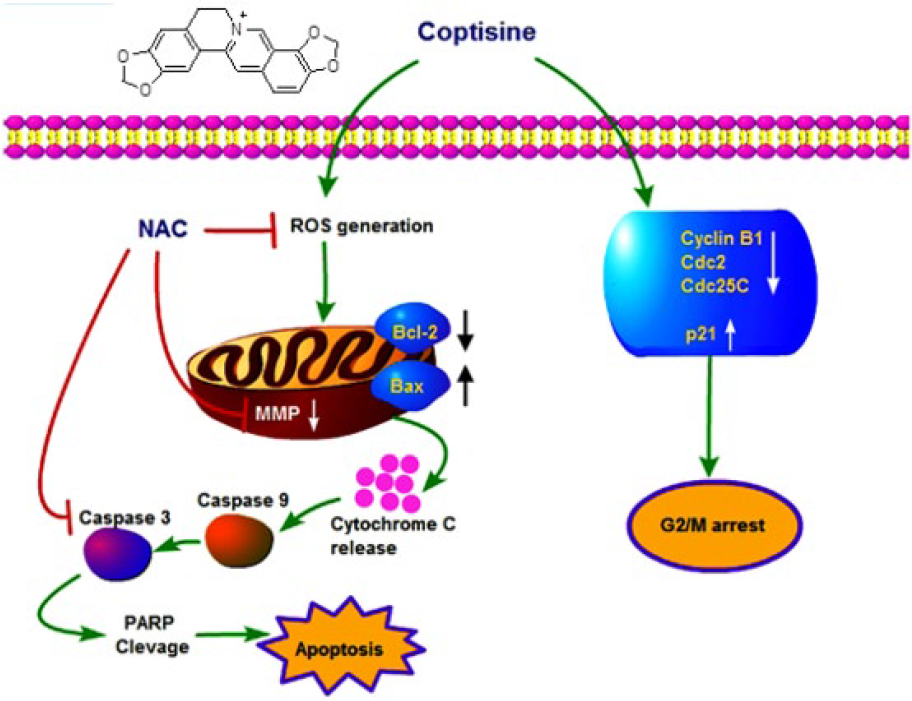
Schematic diagram of the proposed mechanism of COP-induced cytotoxicity.
ROS functions as double-edged sword. ROS at moderate levels can cause genetic mutations, which promote genetic instability, which results in cell proliferation, angiogenesis, and metastasis. All these events allow the cells to survive and subsequently result in cancer development. However, ROS can cause cytotoxicity in cancer cells when it is increased above toxic threshold in response to exogenous agents that increase ROS generation. 55 To ascertain whether COP-induced apoptosis is associated with ROS, COP-treated A549 cells were pre-exposed to the ROS scavenger NAC. The increase in the viability of A549 cells with the addition of NAC, a non-specific antioxidant, suggested the antiproliferative effect mediated through the enhancement of ROS levels. Furthermore, pretreatment with NAC effectively reduced the apoptotic activity, caspase 3 induction, and MMP loss by COP, indicating that COP-induced intrinsic mitochondrial apoptosis was unambiguously mediated by ROS. The study has proved no difference in the mechanism of action between COP and berberine; however the cytotoxicity of COP was observed to be comparatively less, 56 which could be due to the presence of an additional dioxymethylene ring in COP. Although some reports have proved antiproliferative effect of COP, this study is the first proof to validate its effect and mechanism of action against NSCLC (A549 cells).
Conclusion
The study clearly demonstrated that COP inhibited the proliferation of A549 cells through cell cycle arrest and apoptosis. COP-induced DNA damage increased the levels of pH2AX. COP-treated A549 cells were arrested at G2/M phase, accompanied by dowregulation of the expression of cyclin B1, cdc2, and cdc25C and the upregulation of p21. Treatment of A549 cells with COP resulted in the generation of ROS, upregulation of Bax, and downregulation of Bcl-2. The imbalance of Bax and Bcl-2 caused mitochondrial depolarization, which resulted in the cytochrome c release into the cytosol, followed by the activation of caspase 9 and caspase 3, and consequently cleavage of PARP, leading to apoptosis. In a nut-shell, the study concluded that COP-induced mitochondria-dependent apoptosis was mediated through ROS generation. The future focus will be to determine its effect on mouse xenograft model followed by pharmacokinetic studies on both healthy and disease animal models.
Footnotes
Acknowledgements
The authors thank Dr Shrikant Viswanadha, Incozen Therapeutics Pvt. Ltd, Hyderabad, India, for extending laboratory facilities. Authors also acknowledge Research Initiation Grant from BITS-Pilani Hyderabad Campus, Telangana State, India.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Author Poorna Chandra Rao thanks BITS-Pilani Hyderabad Campus for granting Institute Research Fellowship (F.No. 2011PHXF420H).