
Abstract
The methyl methanesulfonate and ultraviolet-sensitive gene clone 81 protein is a structure-specific nuclease that plays important roles in DNA replication and repair. Knockdown of methyl methanesulfonate and ultraviolet-sensitive gene clone 81 has been found to sensitize cancer cells to chemotherapy. However, the underlying molecular mechanism is not well understood. We found that methyl methanesulfonate and ultraviolet-sensitive gene clone 81 was upregulated and the ATM/Chk2 pathway was activated at the same time when MCF-7 cells were treated with cisplatin. By using lentivirus targeting methyl methanesulfonate and ultraviolet-sensitive gene clone 81 gene, we showed that knockdown of methyl methanesulfonate and ultraviolet-sensitive gene clone 81 enhanced cell apoptosis and inhibited cell proliferation in MCF-7 cells under cisplatin treatment. Abrogation of ATM/Chk2 pathway inhibited cell viability in MCF-7 cells in response to cisplatin. Importantly, we revealed that ATM/Chk2 was required for the upregulation of methyl methanesulfonate and ultraviolet-sensitive gene clone 81, and knockdown of methyl methanesulfonate and ultraviolet-sensitive gene clone 81 resulted in inactivation of ATM/Chk2 pathway in response to cisplatin. Meanwhile, knockdown of methyl methanesulfonate and ultraviolet-sensitive gene clone 81 activated the p53/Bcl-2 pathway in response to cisplatin. These data suggest that the ATM/Chk2 may promote the repair of DNA damage caused by cisplatin by sustaining methyl methanesulfonate and ultraviolet-sensitive gene clone 81, and the double-strand breaks generated by methyl methanesulfonate and ultraviolet-sensitive gene clone 81 may activate the ATM/Chk2 pathway in turn, which provide a novel mechanism of how methyl methanesulfonate and ultraviolet-sensitive gene clone 81 modulates DNA damage response and repair.
Keywords
Introduction
Breast cancer is the most common cancer and the leading cause of cancer-related mortality among females worldwide. Almost 1.4 million women were diagnosed with breast cancer worldwide in 2008, and approximately 459,000 deaths were recorded. 1 Many patients with advanced disease become refractory to conventional chemotherapeutic agents, resulting in recurrence and resistance to the treatment.2–4
Cisplatin and its analogues are chemotherapeutic drugs used widely in cancer treatment. Cisplatin can form interstrand crosslinks (ICL), which stall the progression of replication forks during DNA replication.5,6 The DNA adducts formed by cisplatin are converted into DNA double-strand breaks (DSBs). Following DNA damage, cells respond by activating a DNA damage response (DDR) that either leads to repair of the lesion thereby promoting resistance to the drug, or cell death via activation of the apoptotic response. 7
DDR is a signal transduction pathway that coordinates DNA replication, DNA repair, and cell cycle progression and eventually triggers cell death and/or senescence. 8 As a DNA damage sensor, ataxia telangiectasia mutated (ATM) kinase is a nuclear serine–threonine kinase and plays an important role in DDR. ATM initiates the cellular response to DSBs, 9 and is activated through autophosphorylation of the serine 1981 (Ser1981) residue and then activates the distal transducer kinase, Chk2.10–12 It has been shown that loss of ATM or Chk2 expression or inhibition of their kinase activity sensitize cells to death during mitosis. 10
Methyl methanesulfonate and ultraviolet-sensitive gene clone 81 (Mus81) is a highly conserved gene across the species, and is a structure-specific endonuclease with a critical role in the resolution of recombination intermediates during DNA repair after ICL, replication fork collapse, or DSBs.13,14 McPherson et al. 15 demonstrated that both homozygous Mus81−/− and heterozygous Mus81+/− mice exhibited a similar susceptibility to spontaneous chromosomal damage and a predisposition to cancers, which implicate human Mus81 as a candidate tumor suppressor gene. Previous studies also demonstrated that downregulation of Mus81 in hepatocellular carcinoma (HCC), colorectal cancer, astrocytoma, and gastric cancer was closely correlated with the bad progression and prognosis of these malignancies.16–20
Mus81-deficient mice and cells were hypersensitive to the DNA crosslinking agent mitomycin C (MMC). 15 Deleting Mus81 gene could increase the sensitivity of MMC and cisplatin in the human colon cancer cell line HCT116 and HCC.16,21 Our previous studies showed that knockdown of Mus81 by small interfering RNA (siRNA) enhanced the sensitivity to 5-fluorouracil (5-FU) in breast cancer cell lines MCF-7 and T47D. 22 These findings suggest that Mus81 may be the potential marker and target to improve chemosensitivity in human tumors. Although Mus81 expression is correlated with the chemosensitivity in many malignancies, the mechanism is still not clear and needs further exploration.
In this study, MCF-7 cells were used as breast cancer model. Increased chemosensitivity induced by Mus81 knockdown and ATM/Chk2 inhibition in response to cisplatin was investigated. Our results indicate that ATM/Chk2 and Mus81 are mutually activated to control cell sensitivity to cisplatin treatment.
Materials and methods
Cell culture and reagents
Human breast cancer cell line MCF-7 was obtained from the Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, People’s Republic of China). Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; GE Healthcare, Hyclone, MA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, GIBCO, CA, USA) at 37°C in 5% CO2. Cisplatin was purchased from Sigma (St. Louis, MO, USA). ATM inhibitor (KU55933, ab120637) and Chk2 inhibitor (AZD7762, S1532) were provided by Abcam (Cambridge, UK) and Selleck (Houston, TX, USA), respectively. The primary antibodies against Mus81 (ab14387), Bcl-2 (ab7973), phosphorylated ATM (Ser1981) (ab81292), phosphorylated Chk2 (threonine 68 (Thr68)) (ab32148), phosphorylated p53 (Ser15) (ab1431), and β-actin (ab8226) were purchased from Abcam.
Lentivirus-mediated short hairpin RNA
The lentivirus targeting Mus81 was purchased from GeneChem Technology (Shanghai, People’s Republic of China). The sequences of short hairpin RNA (shRNA) targeting Mus81 (confirmed by our previous study 22 ) were as follows: sense: 5′-CCGG GCAGGAGCCATCAAGAATA CTCGAG TATTCTTGATGGCTCCTGC TTTTTG-3′; antisense: 5′-AATTCAAAAA GCAGGAGCCATCAAGAATA CTCGAG TATTCTTGATGGCTCCTGC-3′. And the negative control shRNA sequences were as follows: sense: 5′-CCGG TTCTCCGAACGTGTCACGT CTCGAG TATTCTTGATGGCTCCTGC TTTTTG-3′; antisense: 5′-AATTCAAAAA TTCTCCGAACGTGTCACGT CTCGAG TATTCTTGATGGCTCCTGC-3′. Human MCF-7 cells were seeded in six-well plates at a concentration of 1 × 105 per well and grown overnight. Then, cells were infected with Mus81 shRNA lentiviruses. Medium was changed 8 h after infection.
Cell viability assays
MCF-7 cells were seeded in 96-well plates at a density of 5000 cells per well, and the plates were incubated for 24 h to allow for cell attachment. Then, cells were infected with Mus81 short hairpin RNA (shMus81) lentiviruses. 48 h later, MCF-7 cells were further assembled with cisplatin at concentrations ranging from 0.5 to 16 µg/mL for 24 h. Finally, 10 µL of tetrazolium salt WST-8 (Cell Counting Kit-8 (CCK-8); Keygen, Nanjing, People’s Republic of China) was added to each well (final volume ratio as 10%) for 1 h. The absorbance (OD value) was measured at a wavelength of 450 nm in a spectrometer (Bio-Rad, Fremont, CA, USA). Cell viability was calculated as follows: Viability of cells = Drug-given group OD450/Control group OD450 × 100%.
Cell apoptosis assay
Cell apoptosis rate was analyzed by Annexin V-FITC and propidium iodide (PI) staining using the Annexin V apoptosis detection kit (Keygen) according to the manufacturer’s instructions. Briefly, MCF-7 cells were treated with cisplatin for 24 h at indicated final concentration. Then, cells were removed by trypsinization, rinsed with phosphate buffered saline (PBS), and re-suspended in binding buffer containing Annexin V-FITC and PI for 20 min at room temperature. The samples were analyzed on FC500 flow cytometer.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from MCF-7 cells with TRIzol® (15596026; Thermo Fisher Scientific, Waltham, MA, USA). Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed with the Light-Cycler 480 PCR apparatus (Hoffman-La Roche Ltd, Basel, Switzerland) using SYBR Green PCR master mix (DRR014A; Takara Biotechnology, Dalian, People’s Republic of China). The relative levels of gene expression were calculated by the ΔΔCt method using β-actin as a control and expressed as 2−ΔΔCt. PCR primers of Mus81 and β-actin were used as described previously. 22
Western blot
Cells were lyzed in cold radio immunoprecipitation assay (RIPA) lysis buffer (P0013B; Beyotime, Jiangsu, People’s Republic of China) containing phenylmethylsulfonyl fluoride (PMSF), proteases, and phosphatase inhibitors (P1260; Solarbio, Beijing, People’s Republic of China) on ice. The protein concentrations were determined by the Bicinchoninic Acid assay (Beyotime, Nantong, People’s Republic of China). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Whatman, Maidstone, UK). After non-specific antigen blocking in phosphate buffered saline with Tween 20 (PBS-T) supplemented with 5% bovine serum albumin, the PVDF membranes were incubated with primary antibodies at 4°C overnight. Proteins were visualized on films using secondary horseradish peroxidase (HRP)-conjugated antibodies and ECL-Plus (Thermo Fisher Scientific, New York, NY, USA). β-actin was used as a loading control. The relative protein expression levels were analyzed by Quantity One analysis software.
Statistical analysis
Each experiment was repeated at least three times to confirm that the reliable data were obtained. All data were expressed as mean ± standard error deviation. Statistical significance was tested using one-way analysis of variance (ANOVA) and Student’s t-test. Statistical differences were considered to be significant at p < 0.05.
Results
Cisplatin induces cell apoptosis and reduces cell proliferation in MCF-7 cells
To investigate the pro-apoptosis and cell growth effect of cisplatin, MCF-7 cells were exposed to different concentrations of cisplatin for 24 h. Cell apoptosis was analyzed by flow cytometry with Annexin V/PI staining. Cell viability was evaluated by CCK-8 assay. As shown in Figure 1, the apoptosis efficiencies of cisplatin (at every concentration we tested) in MCF-7 cells were significantly higher than the control group (p < 0.05), and the cell viability rates of cisplatin in MCF-7 cells were significantly lower than the control group (p < 0.05). The results show that cisplatin can effectively induce cell apoptosis and inhibit cell viability in a dose-dependent manner.
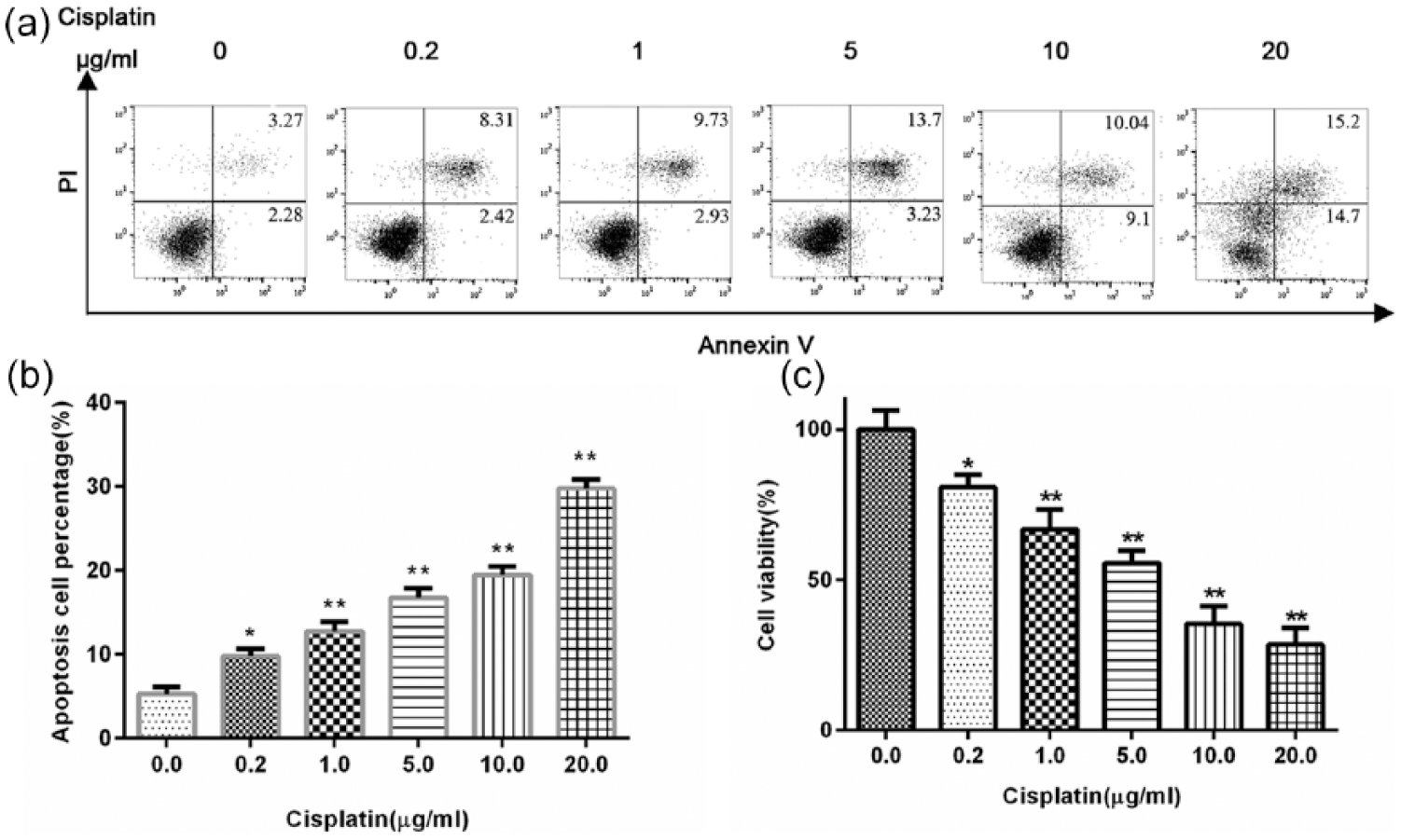
Treatment with cisplatin induces cell apoptosis and inhibits cell viability in MCF-7 cells. MCF-7 cells were treated with different doses of cisplatin for 24 h: (a) cell apoptosis was evaluated by flow cytometry with Annexin V and PI staining, (b) relative cell apoptosis percentage was analyzed, and (c) cell viability was investigated by CCK-8 assay. Data are representative of three different experiments. Statistical analysis was performed comparing treated with untreated cells.
Knockdown of Mus81 sensitizes MCF-7 to cisplatin treatment
MCF-7 cells were infected with lentivirus-mediated shRNA targeting Mus81 gene. The expression of Mus81 was measured by quantitative RT-PCR and western blot 72 h after infection. Compared to the negative control cells, Mus81 mRNA expression level in MCF-7 cells infected with shMus81 lentivirus was remarkably reduced to 25.21% (Figure 2(d)). Western blot analysis also showed that Mus81 protein expression level of MCF-7 cells was significantly decreased after infection of shMus81 lentivirus 72 h later (Figure 2(e)). These results reveal that shMus81 can dramatically reduce the mRNA and protein levels of Mus81 in MCF-7 cells.

Mus81 knockdown sensitizes MCF-7 cells to cisplatin: (a) MCF-7 cells were infected with negative control or shMus81 lentiviruses. 48 h later, 1 µg/mL cisplatin was added for 24 h. Cells were collected and stained with Annexin V and PI. Cell apoptosis was analyzed by flow cytometry, (b) relative MCF-7 cells apoptosis percentage was analyzed, (c) MCF-7 cells were infected with shMus81 lentiviruses for 48 h. Cisplatin with the indicated concentrations (0.5, 2, 4, 8, and16 µg/mL) was added for 24 h. Dose–inhibition rate curves for cisplatin were calculated by CCK-8 assay, (d) MCF-7 cells were infected with shMus81 lentiviruses for 72 h, mRNA was extracted for quantitative RT-PCR analysis, and (e) total proteins were extracted for immunoblotting of Mus81. β-actin served as a loading control.
To determine the impact of Mus81 knockdown on the chemotherapy sensitivity in breast cancer cells, MCF-7 cells were infected with shMus81 lentiviruses for 48 h and then cisplatin was added. 24 h later, cell apoptosis was analyzed by flow cytometry with Annexin V/PI staining. The results showed that Mus81 knockdown remarkably increased cell apoptosis in MCF-7 cells, especially early apoptosis compared to the negative control short hairpin RNA (shCtrl) group (43.7% ± 2.08% vs 15.48% ± 2.67%) (Figure 2(a) and (b)). Cell viability was evaluated by CCK-8 assay, and the results were analyzed by GraphPad Prism 6.0 to establish the dose–inhibition efficiency curves. As shown in Figure 2(c), the inhibition efficiencies of cisplatin in Mus81 knockdown cells were significantly higher than shCtrl group cells (p < 0.01). These findings indicate that Mus81 knockdown can significantly enhance the chemosensitivity of MCF-7 cells in response to cisplatin.
Both ATM and Chk2 inhibitors affect the viability of MCF-7 cells in response to cisplatin
Checkpoint pathways are composed of three main parts: sensors that detect damaged or unreplicated DNA, transducers that relay the signals, and effector proteins that act on the ultimate targets of the checkpoint. As far as we know, all DNA structure checkpoints in mammalian cells require two related kinases, ATM and ataxia telangiectasia and Rad3-related protein (ATR). The signals sensed by ATM and ATR are transmitted through two effector kinases, Chk1 and Chk2, which phosphorylate and thus modify the function of the targets of the checkpoint response.10–12 Previous reports have shown that individual checkpoint abrogators enhance the efficacy of DNA targeting anticancer drugs as well as of radiotherapy. 10 To establish whether this approach is also valid for cisplatin, the influence of ATM (KU55933, 10 µM) and Chk1/Chk2 (AZD7762, 300 nM) inhibitors on the cell viability to cisplatin was determined. MCF-7 cells were pretreated with KU55933 and AZD7762 for 16 h, and a total of different concentrations of cisplatin were added for another 24 h. Cell viability was evaluated by CCK-8 assay. The results showed that both KU55933 (Figure 3(a)) and AZD7762 (Figure 3(b)) could reduce the MCF-7 cells viability compared to the untreated and dimethylsulfoxide (DMSO) treated MCF-7 cells in response to cisplatin (p < 0.05).
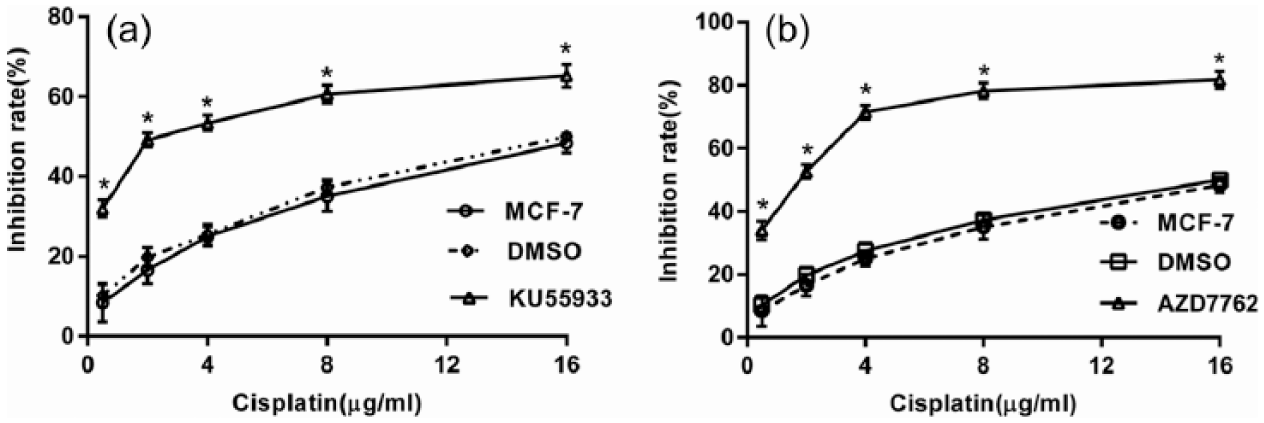
Effects of ATM and Chk2 inhibitors on cell proliferation in response to cisplatin. MCF-7 cells were pretreated with ATM (KU55933, 10 µM) and Chk1/Chk2 (AZD7762, 300 nM) inhibitors for 16 h, then different concentrations of cisplatin (0.5, 2, 4, 8, and16 µg/mL) were added in combination with KU55933 and AZD7762 for 24 h. Cell viability was evaluated by CCK-8 assay: (a) effect of ATM inhibitors on cell proliferation under cisplatin treatment and (b) effect of Chk2 inhibitors on cell proliferation under cisplatin treatment. Statistical analysis was performed comparing treated cells with controlled cells.
Cisplatin treatment upregulates Mus81 expression and activates ATM/Chk2 pathway
DDR is a result of the coordinated actions of DNA damage signaling and repair pathways, cell cycle checkpoints, and apoptosis. To identify the key factors of MCF-7 cells needed for the DDR in response to cisplatin, we first determined the activity of ATM. Western blot was used to determine the activation of ATM, as measured by ATM phosphorylation of Ser1981 after 24 h exposure to five gradient concentrations of cisplatin (Figure 4). The results showed that cisplatin treatments obviously increased ATM phosphorylation in a dose-dependent manner. Coherent with the results for ATM, cisplatin treatment leaded to the activation of Chk2 through phosphorylation on Thr68 (Figure 4). Together, these results indicate that ATM/Chk2 pathway is activated in response to cisplatin.

Treatment with cisplatin activates the ATM/Chk2 pathway and upregulates Mus81 expression in MCF-7 cells: (a) MCF-7 cells were exposed to different concentrations of cisplatin for 24 h. Total proteins were extracted for immunoblotting. β-actin served as a loading control, and (b) relative protein expression levels were analyzed by Quantity One analysis software.
Mus81 is a structure-specific endonuclease that plays an important role in preserving genomic integrity. So we examined the expression of Mus81 in MCF-7 cells after 24h exposure to cisplatin. The results showed that Mus81 expression was significantly elevated after treatment of cisplatin (at every concentration we tested) compared to the control group (p < 0.05) (Figure 4). But the expression of Mus81 was decreased when the cells were treated with cisplatin at concentrations of 8–16 µg/mL compared to 4 µg/mL (p < 0.05).These results indicate that Mus81 is involved in the repair of DNA damage caused by low concentrations of cisplatin treatment.
In addition, we identified the expression of Ser15-phosphorylated p53 and Bcl-2, which was associated with cell apoptosis in MCF-7 cells after cisplatin treatment. As shown in Figure 4, expression of Ser15-phosphorylated p53 was elevated after treatment of cisplatin. Interestingly, Bcl-2 expression was elevated at low concentrations (0.5-2 µg/mL) of cisplatin but decreased at high concentrations (4-16 µg/mL) of cisplatin. These data indicate that treatment with cisplatin in MCF-7 cells induce cell apoptosis, especially at high concentrations.
Cisplatin upregulates Mus81 by activating ATM/Chk2 pathway in MCF-7 cells
Since knockdown of Mus81 and inactivation of ATM/Chk2 pathway inhibit the proliferation and induce cell apoptosis of MCF-7 cells under cisplatin treatment. We asked whether there was some relationship between Mus81 and ATM/Chk2 pathway. Therefore, MCF-7 cells were treated with ATM and Chk2 inhibitors (with 10 µM KU55933 and 300 nM AZD7762, respectively) for 16 h, then cisplatin (1 µg/mL) was added in combination with KU55933 and AZD7762 for 24 h. The results showed that treating MCF-7 cells with KU55933 and AZD7762 could significantly suppress the expression of Ser1981-phosphorylated ATM and Thr68-phosphorylated Chk2, respectively (Figure 5(a)). As shown in Figure 5(a), when the MCF-7 cells were treated with KU55933 and AZD7762, the expression of Mus81 was significantly reduced compared to untreated cells in response to cisplatin. We further analyzed the Mus81 mRNA level of MCF-7 cells in response to cisplatin after being treated with KU55933 and AZD7762. Consistent with the decreased protein levels, both KU55933 and AZD7762 reduced Mus81 mRNA expression (Figure 5(b)). These data indicate that Mus81 is positively controlled by ATM/Chk2 pathway in response to cisplatin.

Cisplatin upregulates Mus81 by activating ATM/Chk2 pathway in MCF-7 cells. MCF-7 cells were pretreated with ATM inhibitor (KU55933, 10 µM) and Chk2 inhibitor (AZD7762, 300 nM) for 16 h. A total of 1 µg/mL cisplatin was added in combination with them for 24 h: (a) total proteins were extracted for immunoblotting to detect pATM-S1981, pChk2-T68, Mus81, and β-actin. β-actin served as a loading control, and (b) total mRNA were extracted for RT-PCR to detect Mus81 mRNA levels.
Mus81 knockdown enhances cisplatin-induced cell death by activating p53 and inhibiting ATM/Chk2 pathway
MCF-7 cells were infected with shMus81 lentiviruses for 48 h and incubated with cisplatin for another 24 h. Total proteins were extracted for immunoblotting. As shown in Figure 6, the expression level of phosphorylated ATM (Ser1981) and phosphorylated Chk2 (Thr68) in MCF-7 cells were significantly decreased in the shMus81 group compared to shCtrl and untreated groups in response to cisplatin. However, the expression of phosphorylated p53 (Ser15) was elevated in the shMus81 group compared to the other groups, and Bcl-2 expression was significantly decreased in response to cisplatin. These results suggest that Mus81 knockdown sensitizes MCF-7 cells to cisplatin by inhibiting ATM/Chk2 pathway and activating p53/bcl-2 pathway.

Mus81 knockdown activates ATM/Chk2 pathway and p53/Bcl-2 in MCF-7 cells under cisplatin treatment. MCF-7 cells were infected with shMus81 lentiviruses for 48 h. A total of 1 µg/mL cisplatin was added for 24 h: (a) total proteins were extracted for immunoblotting to detect pATM-S1981, pChk2-T68, pP53-S15, Bcl-2, Mus81, and β-actin. β-actin served as a loading control, and (b) relative protein expression levels were analyzed by Quantity One analysis software.
Discussion
In this study, we found that knockdown of Mus81 and abrogation of ATM/Chk2 pathway sensitized MCF-7 breast cancer cells to cisplatin treatment; importantly, cisplatin was able to upregulate Mus81 by activating the ATM/Chk2 pathway, which was demonstrated by ATM and Chk2 inhibitors abrogating cisplatin-increased Mus81 expression; interestingly, knockdown of Mus81 sensitized MCF-7 cells to cisplatin by inhibiting ATM/Chk2 pathway and activating p53/bcl-2 pathway, indicating that ATM/Chk2 may regulate Mus81 expression through a positive feedback loop.
ICL-based chemotherapies, such as cisplatin, is the first-line drug for the treatment of many cancers including breast cancer. However, the efficacy of cisplatin is significantly hindered by the development of resistance during treatment. Alterations of DNA damage repair pathway are now recognized as important factors in mediating this situation. ATM/Chk2 pathway plays important roles in DDR. ATM acts as a sensor that detects damaged or unreplicated DNA. The signal sensed by ATM is transmitted through Chk2 which phosphorylates and thus modifies the function of the targets of the checkpoint response. Then, the cell cycle progression is suspended and there is time for cells to repair the damaged DNA. Mus81 is a structure-specific endonuclease that plays an important role in preserving genomic integrity by promoting homologous recombination-mediated repair, mitotic recombination, and the resolution of DNA lesions including ICL.21,23–25 In this study, we observed that cisplatin treatment induced cell apoptosis and inhibited cell proliferation in MCF-7 cells in a dose-dependent manner, indicating cisplatin is a valid agent for treatment of breast cancer. Moreover, cisplatin treatment activated the ATM/Chk2 pathway and increased the expression of Mus81 protein, suggesting that treatment with cisplatin activates the cell cycle checkpoints and DNA repair pathway. Importantly, our observation that MCF-7 cells were sensitized to cisplatin by an ATM/Chk2 kinase inhibitor and shMus81 lentivirus provides strategies for cancer therapy by combination of targeting ATM/Chk2 signaling and downregulation of Mus81 with DNA-damaging modalities. These findings suggest that identifying the alterations of specific proteins and pathways in DNA repair pathways may potentially lead to renewed interest in new markers and novel therapies for cisplatin-resistant cancers.
Recent studies have shown that the forkhead-associated-1 (FHA1) protein docking domain of Cds1 interacts with Mus81,26,27 and Cds1 regulates Mus81 differently in response to replication stress induced by a replication mutant or by hydroxyurea (HU) arrest. 26 Dual inactivation of Chk2 and Mus81 fails to activate G2/M checkpoint in response to MMC treatment and results in mitotic catastrophe and cell death. 28 Since both ATM/Chk2 pathway and Mus81 are involved in DDR and contribute to the cisplatin resistance in MCF-7 cells, we speculate that they may interact with each other. We abrogated the ATM/Chk2 pathway by using KU55933 and AZD7762 in MCF-7 cells under cisplatin treatment. The results showed that the expression of Mus81 was significantly decreased, indicating that ATM/Chk2 upregulates the expression of Mus81 in response to cisplatin. These findings suggest that ATM senses the DNA damage caused by cisplatin, which transmits to Chk2 and then upregulates the Mus81 expression to repair the damaged DNA.
Mus81 is a double-edged sword: In some situations, it is required for maintenance of genomic integrity, but in other circumstances, it can create DNA breaks that threaten genomic stability. Many studies have demonstrated that Mus81 can convert collapsed replication forks into DNA DSBs,29–32 inactivation of Mus81 results in more cell killing and less DSBs. 33 Our previous study has shown that Mus81 knockdown by siRNA could enhance the sensitivity to 5-FU in human MCF-7 and T47D cells. 22 Other studies also indicated that Mus81 was associated with the chemosensitivity in human malignancies such as colon cancer and HCC.16,34 However, the molecular mechanism is largely unclear. In this study, our results demonstrated that Mus81 knockdown by lentivirus-mediated shRNA could remarkably improve the sensitivity to cisplatin in MCF-7 cells. Since ATM/Chk2 pathway is activated by DSBs, we detected the phosphorylation of ATM at Ser1981 and phosphorylation of Chk2 at Thr68 after knockdown of Mus81 in MCF-7 cells under cisplatin treatment. The results displayed that Mus81 deficiency inactivated the ATM/Chk2 pathway. Hence, we propose the speculation that Mus81 cleaves the aberrant replication structures caused by cisplatin and generates DSBs which activate the ATM/Chk2 pathway. But, how Mus81 generates DSBs and activates ATM/Chk2 pathway needs further exploration.
p53 is involved in several functions critical to regulation of cellular growth and death. Apart from apoptosis, p53 is involved in processes like growth arrest and DNA repair as well as senescence. 35 Mus81-deficient mouse embryonic fibroblasts displayed an elevated level of p53 in response to MMC, and p53 inactivation rescued the ICL sensitivity of Mus81-deficient cells at the expense of increased genomic instability. 36 In this study, our results evidenced an elevated expression of phosphorylated p53 in Mus81-suppressed MCF-7 cells after cisplatin treatment, but the Bcl-2 expression changed in the opposite direction. The results were consistent with the recent study in HCC cells. 16 These findings suggest that knockdown of Mus81 activates the p53/Bcl-2 pathway to induce cell apoptosis of MCF-7 cells under the treatment of cisplatin. But how the p53/Bcl-2 pathway is activated by knockdown of Mus81, and whether p53/Bcl-2 is downstream of ATM/Chk2 pathway when the MCF-7 cells are treated with cisplatin should be explored by further research.
In conclusion, we demonstrated that knockdown of Mus81 and abrogation of ATM/Chk2 pathway enhance the chemosensitivity of MCF-7 in response to cisplatin. Mus81 is upregulated by ATM/Chk2 in response to cisplatin, and knockdown of Mus81 by lentivirus-mediated shRNA significantly improves the chemosensitivity of MCF-7 cells by inhibiting the ATM/Chk2 pathway and activating p53/Bcl-2 pathway. Our data provide the basis for the sensitization of breast cancer cells to DNA-damaging agents by targeting Mus81 and ATM/Chk2 pathway.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by the Natural Science Foundation of Zhejiang Province (LY14H200001), Medicines Health Platform Key Project of Zhejiang Province (2013ZDA024), and Medicines Health Platform Project of Zhejiang Province (2015DTA018).