
Abstract
Purpose:
The objective of this study is to determine the impact of exposure to obesity-related systemic factors on fatty acid synthase enzyme (FASN) expression in breast cancer cells.
Methods:
MCF-7 breast cancer cells were exposed to sera from patients having obesity or not having obesity and subjected to quantitative reverse transcription polymerase chain reaction (RT-qPCR). Subsequent MTT and colony-forming assays using both MCF-7 and T-47D cells exposed to sera and treated with or without FASN inhibitor, TVB-3166, were used. MCF-7 cells were then treated with insulin and the sterol regulatory element–binding protein (SREBP) processing inhibitor, betulin, prior to analysis of FASN expression by quantitative RT-qPCR and western blot. Insulin-induced SREBP-FASN promoter binding was analyzed by chromatin immunoprecipitation with an anti-SREBP antibody.
Results:
In response to sera exposure (body mass index [BMI] >30) there was an increase in FASN expression in breast cancer cells. Furthermore, treatment with the FASN inhibitor, TVB-3166, resulted in a decreased breast cancer cell survival and proliferation while increasing apoptosis upon sera exposure (BMI >30). Insulin-exposed MCF-7 cells exhibited an increased FASN messenger RNA and protein expression, which is abrogated upon SREBP inhibition. In addition, insulin exposure induced enhanced SREBP binding to the FASN promoter.
Conclusions:
Our results implicate FASN as a potential mediator of obesity-induced breast cancer aggression and a therapeutic target of patients with obesity-induced breast cancer.
Introduction
At present, obesity is a rapidly rising epidemic 1 with a prevalence of up to 34% in the United States. 2 Moreover, breast cancer is among the highest prevalence of cancer-related death in postmenopausal women with increases commensurate with body mass index (BMI).3,4 With obesity rates at epidemic proportions, uncovering mechanisms of obesity-induced breast cancer progression is of utmost importance. There are multiple proposed mechanisms by which obesity can lead to enhanced tumorigenesis, such as elevated circulating free fatty acids (FFAs) and lipid signaling, inflammation, adipokine secretion, and the bioavailability of growth factors and hormones, such as insulin, insulin-like growth factor-1 (IGF-1), and estrogens. 5
A well-established hallmark of cancer includes the alteration in metabolism to sustain proliferative capacity and synthesize essential biomolecules. 6 Of these metabolic alterations commonly seen in cancers, the enhancement of de novo lipogenesis enables cancer cells to synthesize fatty acids from carbohydrate or amino acid sources, such as glucose and glutamine, respectively.6,7 Although the primary function of de novo lipogenesis in liver and adipose tissue is to esterify fatty acids into triglycerides for fat storage in response to excess caloric intake, cancer cells use fatty acids for esterification into phospholipids for membrane synthesis and lipid raft incorporation, β-oxidation for energy, and substrates for post-translational acylation of proteins.7,8 Although most quiescent cells acquire their fatty acids through diet, the high metabolic demand of cancers and other highly proliferative tissues, such as lactating breast and cycling endometrium, make it necessary to acquire an aberrant lipogenic programming. 8 The process of de novo lipogenesis involves several enzymatic steps beginning with the ATP citrate lyase (ACLY) catalytic conversion of citrate and coenzyme-A (CoA) to acetyl-CoA and oxaloacetate in the cytosol, which is followed by the carboxylation of acetyl-CoA to malonyl-CoA, catalyzed by the acetyl-CoA carboxylase (ACC) enzyme. Acetyl-CoA acts as the initial carbon donor, while malonyl-CoA is repeatedly added to the carbon chain through multiple cycles of chain elongation in the fatty acid synthase enzyme (FASN) complex, which results in the final 16-carbon fatty acid product, palmitate.7,8 Protein kinase B (Akt) and mammalian target of rapamycin complex 1 (mTORC1) are central mediators of metabolic signaling, where they receive upstream activation from insulin and insulin-like growth factor receptors and transmit them to downstream targets for cell growth and proliferation through lipogenesis and protein synthesis. Previous studies have illustrated Akt and mTORC1 to be regulators of transcription for de novo lipogenesis through both sterol regulatory element–binding protein-1 (SREBP-1) dependent and independent pathways.9-11 Moreover, inhibition of mTORC1 using rapamycin and Torin1 complex inhibitors has shown an attenuation of messenger RNA (mRNA) expression for enzymes controlling de novo lipogenesis such as ACLY and FASN.9,11
Since the discovery of FASN as an oncogenic antigen overexpressed in aggressive breast cancers, preclinical and clinical research for targeted therapies has been extremely active. 12 However, early FASN inhibitors were limited in their use as therapeutic agents for cancer patients because they induced cachexia, 13 which is detrimental to cancer patients undergoing treatment. Newer, more targeted inhibitors toward FASN have shown promise in early clinical studies, as they have not shown signs of causing significant weight loss or other severe toxicities. 14 Currently, TVB-2640, a FASN inhibitor that targets the ketoacyl reductase domain, is in phase II of clinical trials for patients heavily pretreated with paclitaxel with minimal side effects. 14 There are multiple proposed mechanisms by which FASN inhibitors promote cancer cell apoptosis including the reduction of palmitate for post-translational protein modifications and membrane composition, toxic accumulation of the FASN substrate malonyl-CoA, decreased membrane synthesis, and disruption of lipid rafts.6,13,15-18 In an interesting study using the novel FASN inhibitor, TVB 3567, which tracked tumor metabolism by isotopically labeled [U-13C] glucose incorporation into [13C] C16:0 palmitate; Daniel Benjamin and colleagues found that 231-MFP and MCF-7 breast cancer cells not only decreased the de novo glucose-derived palmitate synthesis but also exhibited decreased cell viability in response to FASN inhibition. 16
We have previously demonstrated increased circulating palmitate in obese patients with breast cancer and obesity-associated factors to induce a more aggressive phenotype in luminal A breast cancer cells.4,19,20 Thus, this study investigates the role of obesity in mediating FASN expression in luminal A breast cancer cells.
Methods
Serum samples
All serum collection processes were approved by the institutional review board (IRB) and consent was given. Serum was collected from postmenopausal women. Body mass index was calculated and serum was pooled according to the BMI of the patient based on BMI categories. Colony formation assays were conducted using sera as previously described. 19 All other data were collected using serum samples collected from postmenopausal women waiting to go on trial. One patient was considered non-obese (BMI = 18.5 kg/m2) and the other patient was considered obese (BMI = 31 kg/m2). Sera were analyzed as previously indicated, 19 examining IGF-1, leptin, and IL-6 levels for consistency.
Cell culture
MCF-7 and T-47D human breast cancer cell lines were maintained in Improved Minimum Essential Medium (IMEM) (GIBCO Life Technologies, Grand Island, New York) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a humidified 37°C incubator.
Quantitative RT-PCR
Fatty acid synthase enzyme mRNA levels in the MCF-7 and ASC cell lines were quantified after 24-hour exposure to 2% (BMI = 30) or (BMI = 18.5) sera in serum-free media (SFM). Total RNA was isolated using TRIzol reagent (Invitrogen) and reverse transcribed using Promega’s ImProm II Reverse Transcription System. QuantiFast SYBR Green PCR kit was used, per the manufacturer’s instructions. The data shown represent the average of 3 independent experiments. All values were standardized to their Ct values of the actin housekeeping gene. Data are representative of fold or percent.
The following primers were used for the experiment:
FASN:

Actin:
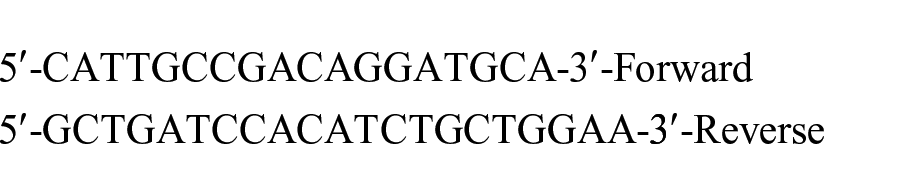
Colony-forming assay
MCF-7 and T47-D were seeded in complete IMEM at a density of 500 cells per well in a single cell suspension in a 12-well plate. After 24-hour incubation and phosphate-buffered saline (PBS) wash, the cells were exposed to 2% sera (BMI = 18.5 or BMI >30) + 8% FBS with or without TVB-3166. Medium was replaced every third day. On day 10 of incubation with the indicated treatments, following a PBS wash, 2% crystal violet in methanol was added to fix and stain the cells. Groups of 50 or more cells were counted as colonies.
MTT assay
MCF-7 cells were seeded in IMEM supplemented with 10% FBS at a density of 5 × 103 in 96-well plates. After a 24-hour incubation, the cells were exposed 2% sera (BMI 18.5 or BMI >30) + 8% FBS with or without TVB-3166 (FASN Inhibitor) for 96 hours. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT reagent) was purchased from Sigma-Aldrich (St. Louis, Missouri). MTT reagent in PBS (5 mg/mL) was then added to each well. After 2 hours of incubation at 37°C, the media were removed and 100 µL dimethyl sulfoxide (DMSO) added to each well. Absorbance was read at 570 nm on a spectrometer. Relative cell inhibition was calculated by dividing each absorbance value by the absorbance for cells grown in corresponding sera without TVB-3166. Data shown represent the average of 3 independent experiments.
Western blotting
MCF-7 cells were seeded in 6-well dishes and serum starved for 16 hours prior to exposure with either sera from patients with obesity or without obesity or insulin (100 nM) for 24 hours with or without betulin (13.5 μM) treatment. Cells were washed with 1× PBS and lysed with cell lysis buffer (buffer [1% SDS, 1 mM EDTA, 10 mM Tris-HCL pH 8.0] supplemented with Halt phosphatase and protease inhibitor cocktail kit [Thermo Fisher, #787429]). Lysates were subjected to sonication using a Misonix ultrasonic liquid processor. Lysates were then centrifuged 10 000 r/min at 4°C for 10 minutes. The resulting supernatant was collected and 10 µL subjected to Pierce BCA protein assay (Thermo Fisher #23227). Then 50 µg of protein were solubilized in 4× loading buffer (8% SDS, 250 mM Tris-HCL [pH 6.8], 20% β-mercaptoethanol, 40% glycerol, and 0.2% bromophenol blue) and heated to 95°C for 5 minutes. Lysates were resolved on an 8% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and transferred to a nitrocellulose membrane for 1 hour. Membranes were blocked in 5% milk in Tris-Buffered Saline + Tween 20 (TBST) for 1 hour at room temperature and washed with TBST for 3 times for 5 minutes. Primary antibodies in dilution buffer—FASN (1:1000), Cell Signaling Technologies, #3180; GAPDH (1:300), Santa Cruz Biotechnology, #SC-2577; SREBP (1:300), Santa Cruz Biotechnology, #SC-366; PARP (1:100), Cell Signaling Technologies, #9532—were incubated at 4°C overnight. Membranes were washed the following day with TBST once for 15 minutes followed by an additional 5-minute wash. Secondary antibodies anti-rabbit HRP-linked IgG (CST, #7074) (1:3000) or anti-mouse HRP-linked IgG (CST, #7076) (1:5000) were diluted in 5% milk and added to the membranes for 1 hour at room temperature. Membranes were washed in TBST 3 times for 5 minutes and incubated with West ATTO Chemiluminescent substrate (Thermo Fisher, #A38554) prior to being visualized with a Syngene imaging system using the gene snap technology program. Bands were quantified using Image J software and standardized to the GAPDH loading control.
Chromatin immunoprecipitation assay
MCF-7 cells were grown until 70% confluency in 150 mm dishes and treated with 100 nM human recombinant insulin with or without the SREBP processing inhibitor, betulinic acid (EMD Millipore, #569371) for 3 hours. Cells were crosslinked with 1% formaldehyde for 10 minutes after which the reaction was terminated by the addition of glycine at 0.125 M. Cells were lysed in sucrose cell and nuclear lysis buffer (cell lysis buffer: Tris-HCl, 1% Triton-X-100, and 30% sucrose, nuclear lysis buffer: Tris-HCl and 30% sucrose) followed by chromatin digestion with a micrococcal nuclease enzyme per manufacturer’s instructions (Cell Signaling Technologies, #10011). Chromatin was sheared using a VirSonic 100 ultrasonic sonicator 3× 20 seconds on ice. The resulting sheared chromatin was precleared by protein G agarose (Santa Cruz Biotechnology, SC-2002) for 30 minutes at 4°C. The supernatant was diluted 5-fold in chromatin immunoprecipitation buffer. 10 µg of chromatin-protein complex was then subjected to immunoprecipitation with 2 µg of anti-SREBP (Santa Cruz Biotechnology, sc-366) or 2 µg of normal rabbit IgG (Cell Signaling Technologies, #2729P) overnight with rotation at 4°C. Meanwhile, 50 µL of protein G agarose beads was washed in dilution buffer and added to each immunoprecipitation for 2 hours at 4°C. Beads were then washed 4 times with low-salt wash buffer (0.1% SDS, 1% Triton-X-100, 2 mM EDTA, 20 mM Tris pH 8.0, and 150 mM NaCl) and once with a high-salt buffer (0.1% SDS, 1% Triton-X-100, 2 mM EDTA, 20 mM Tris pH 8.0, and 500 mM NaCl). Beads were eluted at 65°C in elution buffer (1% SDS, 0.1 M NaHCO3). The supernatant was digested with proteinase K and crosslinks reversed by heating at 65°C for an additional 2 hours. DNA was isolated per manufacturer’s protocol using DNA purification spin columns (New England Biolabs, T3010S). Purified DNA was then subjected to quantitative polymerase chain reaction (qPCR) amplification with the following FASN promoter-specific primers. 21
FASN promoter-specific primers:

The resulting DNA and primers were used with SYBR Green Power Track master mix as per manufacturer’s instructions (Thermo Fisher). Fold enrichment was used to quantify the resulting Ct values. Ct values were normalized against their respected negative control antibody (Ct SREBP—Ct IgG rabbit). Subsequent values were enriched using the (2−ΔΔCT) formula.
Statistical analysis
The Student t-test was used to analyze the differences between conditions. Two-way analysis of variance (ANOVA) followed by Tukey Honest Significant Difference post hoc analysis was performed for analysis of multiple conditions. Collected data were considered statistically significant at a P value less than .05.
Results
Obesity promotes FASN expression in breast cancer cells
De novo lipogenesis is now considered a hallmark for many cancers including that of breast and FASN is a major contributor to endogenous lipogenesis and breast tumor progression and aggressiveness. To investigate the effects of FASN expression as a contributor to obesity-induced breast cancer, sera was obtained from postmenopausal women who had obesity or did not have obesity and exposed to MCF-7 breast cancer cells. Upon sera exposure from patients who had obesity (BMI >30), there was a significant (P = .034) increase in FASN expression in MCF-7 cells exposed to sera from patients with obesity (BMI >30) compared with that from patients who did not have obesity (BMI = 18.5) (Figure 1A). To demonstrate whether obesity also affected FASN protein expression in breast cancer, we exposed MCF-7 cells to the same conditions for 24 hours and subjected them to western blot analysis. Obese sera exposure greatly increased FASN protein expression in MCF-7 breast cancer cells (Figure 1B). These results illustrate a potential mechanism in which obesity contributes to breast cancer progression through FASN.

(A). FASN is overexpressed in exposed breast cancer cells with sera from patients with obesity. (A) MCF-7 breast cells were exposed to either sera from patients with obesity (BMI >30) or patients who did not have obesity (BMI = 18.5-24.9) for 24 hours and then subjected to quantitative reverse transcription polymerase chain reaction. The exposure to sera from patients with obesity for 24 hours resulted in a significant-fold increase (P < .05) in FASN mRNA in MCF-7 breast cancer cells compared with patients who did not have obesity. The results are represented as means (±SD) of 3 independent trials; n = 3. Each trial was performed in triplicates. (B) Sera exposed cells were subjected to western blot analysis for FASN and GAPDH. Numbers under the western blot represent an average band intensity of FASN/GAPDH from 3 biological triplicates (n = 3).
FASN promotes breast cancer cell survival and proliferation
Fatty acid synthase inhibition has been an active therapeutic target for various cancers and mediates its effects through numerous mechanisms. To investigate the effects of FASN expression and breast cancer cell survival in obese conditions, MCF-7 cells were exposed to sera from patients having or not having obesity and treated with the FASN inhibitor, TVB-3166 or DMSO for 10 days. Subsequent cell survival was analyzed using a colony-forming assay. The results indicate that treatment with the FASN inhibitor TVB-3166 reduced survival upon sera exposure from patients with obesity in both MCF-7 and T-47D lines (Figure 2A and B). These results appear consistent with previous studies demonstrating the impact of FASN inhibition on breast cancer cell survival and highlight the potential for targeted therapy for breast cancer in obese individuals.

(A and B) FASN contributes to obesity-induced breast cancer cell survival. (A) MCF-7 breast cancer cells were exposed to either sera from patients with or without obesity and drug vehicle Dimethyl Sulfoxide (DMSO) or FASN inhibitor (TVB) 50 nM for 10 days. Cell colonies were counted in response to treatments. Graphs represent an average of at least 3 trials (n = 3) of surviving colony counts in MCF-7 breast cancer cells. Each trial was performed in triplicate. Data are represented as a mean ± SD of 3 independent experiments. * indicates statistical significance (P = .049) between BMI (>30) and DMSO versus TVB treatment. “NS” denotes there was no significant difference between DMSO versus TVB upon exposure from BMI (18.5-24.9) sera. (B) T47-D breast cancer cells were exposed to sera from patients with or without obesity (BMI = 18.5-24.9) or (BMI >30) and drug vehicle (DMSO) or FASN inhibitor (TVB) for 24 hours. Cell colonies were counted in response to treatments. Data represent a mean ± SD of 3 independent trials (n = 3) of surviving colony counts in T47-D cells. Each trial was performed in triplicate.
FASN inhibition reduces obesity-induced breast cancer cell proliferation
To investigate whether the FASN inhibitor would be a more targeted option for obesity-induced breast cancer, breast cancer cells were exposed to sera from patients having or not having obesity and treated with FASN inhibitor TVB-3166 at various concentrations for 96 hours. Cell proliferation upon treatment with differing concentrations of the FASN inhibitor, TVB-3166, was analyzed using an MTT assay. There was a reported decrease in cell proliferative response to treatment of TVB-3166 upon sera exposure from patients with obesity compared with patients who did not have obesity (Figure 3A). Upon increasing concentrations of TVB-3166, there was a stepwise decrease in cell viability with obese sera exposed that was not apparent in non-obese sera-exposed breast cancer cells. Together, these results show that FASN inhibition using TVB-3166 at 150 nM has an effect on obese sera-exposed breast cancer cell proliferation, which was not observed in non-obese sera-exposed cells. To determine whether this decreased proliferation was due to apoptosis, we exposed breast cancer cells to either obese or non-obese sera along with 150 nM of TVB-3166 for 96 hours. Treatment of obese sera-exposed breast cancer cells resulted in significantly increased cleaved PARP, a marker of apoptosis, compared with cells exposed to non-obese sera (Figure 3B). Given these results, FASN inhibition could offer a selective treatment option for obesity-induced breast cancer.

(A and B) FASN inhibition reduces obese-exposed breast cancer cell proliferation. (A) MCF-7 cells were exposed to 8% FBS with 2% sera either from patients with obesity or from patients without obesity for 24 hours and treated with 0, 50, 100, or 150 nM of the FASN inhibitor TVB-3166. Graphs indicate a mean fold change to 0 nM TVB-3166 of 3 independent trials ± SEM (n = 3). Each trial was performed in triplicate. Statistical analysis was performed by 2-way ANOVA followed by a Tukey Honest Significant Difference post hoc test. Statistical codes are as follows” “*” = P ⩽ .05, “**” = P ⩽ .01. (B) MCF-7 cells were exposed to media with 8% FBS and 2% sera from patients with (BMI >30) or without obesity (BMI = 18.5) along with either Dimethyl Sulfoxide (DMSO) or 150 nM TVB-3166 for 72 hours. Cells were subjected to western blot analysis for PARP and GAPDH. The graph below represents an average band intensity from 3 independent trials n = 3 ± SD of cleaved-PARP/total. The Student t test was performed for statistical analysis.
Insulin-induced SREBP binding to FASN promoter
Obesity is associated with metabolic perturbations such as insulin resistance and subsequent increases in circulating insulin, FFAs, and hormones.22,23 Downstream effectors from insulin and insulin-like growth factor-1 receptor (IGF-1R) include the canonical phosphatidylinositol-3k kinase (PI3K)-Akt-mTORC1 pathway that can result in an increase stabilization and proteolytic processing of SREBPs, which can bind to sterol response elements on the FASN promoter and facilitate transcription. 24 Thus, we aimed to investigate if insulin was the mediator of the increased FASN expression observed in breast cancer cells exposed to sera from patients with obesity. To investigate the role of insulin as a mediator of obesity-induced FASN expression, MCF-7 breast cancer cells were treated with insulin with or without betulin, the SREBP processing inhibitor, for 3 hours before being subjected to RT-qPCR. Inhibition of SREBP processing by betulin treatment resulted in a significant decrease in insulin-induced FASN expression in MCF-7 cells (Figure 4A). In addition, insulin resulted in an increase in FASN protein level, which was attenuated upon SREBP inhibition by betulin treatment (Figure 4B). To determine a specific insulin-dependent mechanism of FASN regulation in breast cancer, MCF-7 cells were exposed to insulin with or without betulin and crosslinked. Crosslinked cells were subjected to chromatin immunoprecipitation with SREBP or negative IgG control antibodies and amplified with an FASN SRE-binding site-specific primer (Figure 4B). 21 Exposure to insulin resulted in a significant-fold enrichment in the FASN promoter (P = .036) (Figure 4C). In addition, the prior exposure to SREBP processing inhibitor, betulin, abrogated the promoter enrichment in response to insulin exposure (Figure 4C). Thus, circulating insulin concentrations in obesity could be a key mediator of metabolic reprogramming in obesity-induced breast cancer through the SREBP-FASN pathway.

(A-C). Insulin promotes FASN expression through the binding of SREBP to the FASN gene promoter. (A) MCF-7 cells were exposed to serum-free media alone, and serum-free media exposure to insulin (100 nM) with or without SREBP processing inhibitor, betulin (13.5 μM). RNA was isolated and subjected to quantitative reverse transcription polymerase chain reaction analysis for FASN mRNA expression. mRNA expression for FASN in each condition was standardized to Actin. The standardized values were then made relative to the serum-free media alone condition and expressed as relative percent expression of FASN. Bars represent a mean ± SD of 3 independent trials (n = 3). Each trial was performed in triplicate. (B) MCF-7 cells were serum starved for 16 hours prior to exposure to complete growth medium alone, with 100 nM of insulin, or 100 nM of insulin with 13.5 μM of betulin for 24 hours before protein was harvested. FASN and SREBP protein were detected by western blotting. GAPDH served as the internal loading control. Experiments were repeated in biological triplicate (n = 3). (C) Schematic of the region SRE/E-box binding region of FASN promoter used to amplify DNA loci. (D) MCF-7 cells were exposed to insulin (100 nM) with or without an SREBP processing inhibitor (betulin 13.5 μM) for 3 hours prior to crosslinking SREBP to SRE promoter regions of FASN gene. Chromatin immunoprecipitation was performed with either anti-SREBP or anti-rabbit control overnight. Bars represent a mean ± SD fold enrichment of FASN promoter region relative to the respected conditions. N.S. indicates no statistical significance between groups. The Student t test was performed to test for significance in (A, C).
Discussion
Obesity-induced breast cancer confers a treatment-resistant and aggressive phenotype. Therefore, identifying the mechanisms of how obesity confers this phenotypic switch in breast cancer is of utmost importance. In this study, we demonstrated a potential link between obesity and increased breast cancer aggression through FASN expression. Increased FASN expression was observed in cancer cells exposed to sera from patients with obesity compared with those from patients who did not have obesity.
Moreover, obesity is often accompanied by a concomitant hyperinsulinemia brought on by insulin resistance.25,26 The increased circulating growth factors such as insulin and IGF-1 can result in signaling for cell survival, growth, proliferation, differentiation, and migration through the insulin receptor (INSR), IGF-1R, or the hybrid INSR/IGF1R heterodimer complex.25,26 In addition, IGF-1 has been documented as being more bioavailable in obese subjects due to various proposed mechanisms including hyperinsulinemia and its effects of inhibiting the synthesis of insulin-like growth factor binding proteins (IGFBPs) 1, 2, and 3 in the liver.25,26 Downstream effector proteins of INSR and IGF-1R include the metabolic regulators, Akt and mTORC1.9-11 The classical mechanism of Akt and mTORC1 transcriptional control of lipid synthesis is through the family of master transcriptional regulators of both cholesterol and fatty acid biosynthesis, SREBPs. The SREBPs have been extensively studied as being transcription factor that binds to the promotor regions of enzymes that control both endogenous cholesterol and fatty acid synthesis, 3-hydroxy-3-methyl-glutryl (HMG)-CoA reductase and FASN, respectively.9,10,27 Some established mechanisms of FASN regulation include Akt inhibition of the insulin-induced gene product (Insig) resulting in pre-SREBP being cleaved in the endoplasmic reticulum by SREBP cleavage-activating protein (SCAP) and inhibition of glycogen synthase kinase 3β (GSK-3β) resulting in protection of SREBP from ubiquitin-mediated proteasomal degradation, and it is well established that SREBPs are one of the master regulators of FASN expression and that they have recognition elements on FASN promoter genes. 27 Thus, the resulting overexpression of FASN in sera-exposed MCF-7 cells from patients with obesity could be explained by downstream signaling from Akt and mTORC1.
Furthermore, we illustrated a decrease in breast cancer cell survival and proliferation in response to FASN inhibition by TVB-3166. There are multiple reported effects of attenuating cancer cell survival by the use of FASN inhibitors such as toxic levels of the FASN substrate malonyl-CoA, partitioning lipid rafts, decreasing palmitate for subsequent protein palmitoylation, and a decreased fatty acid storage for β-oxidation.6,7,28 A mechanistic observation of insulin-induced SREBP binding to the FASN promoter was observed using chromatin immunoprecipitation, and this was ameliorated by pre-treatment with betulin, naturally derived product that acts to inhibit SREBP processing through the inhibition of SCAP in the endoplasmic reticulum (EnR). 29 Though there have been some associative studies with insulin and the SCAP-sequestering Insig, further investigation in the role of Insig in obesity-induced FASN expression is warranted. Given that an SCAP inhibitor can hamper the insulin-induced increase in FASN expression, the Insig-SCAP complex in the EnR could play a central role in the insulin-FASN connection. Also, additional investigations should address other potential roles of both Akt and mTORC1 in mediating FASN programming in cancer.
In summary, FASN that was illustrated as a potential mediator of obesity-induced breast cancer aggression may be more effectively targeted in the patients who have obesity. Based on the results, FASN shows promise as a novel, nontoxic, therapeutic target for the breast cancer patient with obesity
Footnotes
Acknowledgements
The authors thank the research and oncology nurses, Kalya DiMarco and Christine Maglaki (UT Health San Antonio) for obtaining the sera and the patients for allowing this study to be possible. They also thank Dr Andrew Brenner (UT Health San Antonio) for kindly providing the sera and Michael Garcia (UT Health San Antonio) for facilitating the process.