
Abstract
Since the incidence of breast cancer increases dramatically all over the world, the search for effective treatment is an urgent need. Metformin has demonstrated anti-tumorigenic effect both in vivo and in vitro in different cancer types. This work was designed to examine on molecular level the mode of action of metformin in mice bearing solid Ehrlich carcinoma and to evaluate the use of metformin in conjunction with doxorubicin as a combined therapy against solid Ehrlich carcinoma. Ehrlich ascites carcinoma cells were inoculated in 60 female mice as a model of breast cancer. The mice were divided into four equal groups: Control tumor, metformin, doxorubicin, and co-treatment. Metformin (15 mg/kg) and doxorubicin (4 mg/kg) were given intraperitoneally (i.p.) for four cycles every 5 days starting on day 12 of inoculation. The anti-tumorigenic effect of metformin was mediated by enhancement of adenosine monophosphate protein kinase activity and elevation of P53 protein as well as the suppression of nuclear factor-kappa B, DNA contents, and cyclin D1 gene expression. Metformin and doxorubicin mono-treatments exhibited opposing action regarding cyclin D1 gene expression, phosphorylated adenosine monophosphate protein kinase, and nuclear factor-kappa B levels. Co-treatment markedly decreased tumor volume, increased survival rate, and improved other parameters compared to doxorubicin group. In parallel, the histopathological findings demonstrated enhanced apoptosis and absence of necrosis in tumor tissue of co-treatment group. Metformin proved chemotherapeutic effect which could be mediated by the activation of adenosine monophosphate protein kinase and related pathways. Combining metformin and doxorubicin, which exhibited different mechanisms of action, produced greater efficacy as anticancer therapeutic regimen.
Introduction
Breast cancer is the most common cancer among women and the most likely cause of death in women worldwide as reported by the International Agency for Research on Cancer. 1 The incidence rate of breast cancer varies dramatically across the globe, being always less than 30/105 of women annually in regions like Eastern Asia, compared with more than 90 new cases/105 of women annually in more developed regions, namely, North America and Western Europe. 1 According to the National Population-Based Cancer Registry Program of Egypt, breast cancer threatened about 32.04% of women in Egypt between 2008 and 2011. 2
Metformin (MET) is a biguanine compound that is characterized by its glucose-lowering effect. MET inhibits hepatic gluconeogenesis and stimulates insulin sensitivity and glucose uptake in diabetic patients. 3 MET can inhibit mitochondrial oxidative phosphorylation that leads to an adenosine triphosphate/adenosine monophosphate (ATP/AMP) imbalance and activation of the liver kinase B1–AMP-activated protein kinase (LKB1-AMPK) pathway. 4
The anticancer effects of MET are related to both direct (insulin-independent) and indirect (insulin-dependent) actions of the drug. The direct effects are associated with LKB1-mediated activation of AMPK and a reduction in mammalian target of rapamycin (mTOR) signaling and protein synthesis in cancer cells. The indirect effects originate from its insulin-lowering action since insulin has mitogenic and prosurvival effects. 5
The cellular energy sensor and regulator AMPK play a key role in the regulation of protein and lipid metabolism in response to changes in fuel availability.6,7 Several evidences have demonstrated that AMPK inhibits the anabolic pathways that promote cell growth, such as synthesis of fatty acid, phospholipid, protein, and ribosomal RNA synthesis. 8 Since tumor cells require great demand of energy because of their ability to rapidly grow and divide, it is not surprising that AMPK antagonizes cancer cell growth. 9
Doxorubicin (DOX) is an anthracyclin drug that is used in the treatment of many types of cancers including breast cancer. The anticancer effect of DOX is mainly due to the intercalation into DNA and the disruption of topoisomerase II–mediated DNA repair. Also, it generates free radicals that cause damage to cellular membranes, DNA, and proteins. 10 Cardiotoxicity and myelosuppression are the major systemic toxicities of DOX due to its oxidative stress action, so it is preferred to combine it with other compounds to reduce its dosage without affecting its efficacy. 11
The combination of chemotherapeutic agents with different mechanisms proved higher efficacy in cancer treatment. Therefore, we aimed in this study to identify some of the molecular mechanisms of MET and to demonstrate its possible beneficial role as an adjuvant therapy to DOX for treatment of mammary carcinoma in mice.
Materials and methods
Drugs
DOX hydrochloride (Adriblastina® vials) was obtained from Pharmacia Italia S.P.A. (Milan, Italy). MET hydrochloride was a gift from Amoun Pharmaceutical Company S.A.E. (El Obour City, Cairo, Egypt).
Animals and experimental design
The study was performed in accordance with the guidelines for the care and use of laboratory animals approved by Research Ethical Committee, Faculty of Pharmacy, Tanta University, Egypt (FPTU-REC, 115/2013/620). A total of 60 female Swiss albino mice aged 6–8 weeks and weighed 18–22 g were purchased from National Research Center (Cairo, Egypt). Mice were maintained 1 week for acclimatization in wire cages at identical conditions and provided with standard pellet diet and water ad libitum.
Ehrlich ascites carcinoma (EAC) is derived from mouse breast adenocarcinoma that is aggressive and with a high grade of malignancy.12,13 A fixed number of EAC cells (1 × 106 cells) was obtained from the Pharmacology and Experimental Oncology Unit of the National Cancer Institute (Cairo University, Giza, Egypt). EAC cells were implanted into the peritoneal cavity of a mouse and were allowed to multiply. An ascitic fluid containing Ehrlich tumor cells was developed within 10 days, withdrawn by a sterile syringe, then diluted in 0.9% sterile saline (1:9 v/v), and counted using a Neubauer Hemocytometer (Sigma Aldrich, St. Louis, MO, USA). The cell viability was determined by trypan blue dye exclusion method 14 and was found to be more than 99%.
Solid Ehrlich carcinoma (SEC) was induced by injecting viable EAC cells (1 × 106 cells) subcutaneously into the right thigh of the lower limb of mice. A palpable solid tumor mass (about 100 mm3) was developed within 12 days, after which SEC-bearing mice were randomly divided into four groups, each of 15 mice, as follows: control tumor group was given the vehicle, MET group was given MET 15 mg/kg, 15 DOX group was given DOX 4 mg/kg, 16 and co-treatment group was given both MET and DOX at the specified doses. Each of MET and DOX were injected i.p. for 4 cycles every 5 days (on the 12th, 17th, 22nd, and 27th days).
The survival rate was calculated for each experimental group as follows: survival rate = (number of live animals in a group on the 28th day/number of animals in the same group at the start of experiment) × 100. 17 The dimensions of the tumor were measured with a vernier caliper (Tricle Brand, Shanghai, China) on the 12th day and then day after day till the end of experiment. Tumor volume was calculated using the following formula: tumor volume (mm3) = 0.52 AB2, where A is the length of minor axis and B is the length of major axis. 18 Tumor inhibition rate (TIR) was calculated according to Salem et al. 19 using the following formula: TIR = ((mean tumor volume of control tumor group − mean tumor volume of treated group) × 100/mean tumor volume of control tumor group).
On the 28th day, mice were anesthetized by ether, blood was withdrawn via cardiac puncture, and then mice were killed by cervical dislocation. Serum samples were obtained by centrifugation at 3000 r/min for 20 min using cooling centrifuge (Laborzentrifugen 3-3OK; Sigma, Munich, Germany) and stored at −80°C. Tumor tissue was carefully excised and divided into portions. One portion was fixed in 10% formalin for histopathological examination, and the other portions were kept frozen at −80°C.
Assessment of biochemical parameters
Enzyme-linked immunosorbent assay (ELISA) kits were obtained from Glory Science Co. (Del Rio, USA) and utilized to determine the concentration of the tumor suppressor gene P53 in serum and phosphorylated adenosine monophosphate protein kinase (PAMPK)-pT172 and the nuclear factor-κB (NF-κB)-p65 in tumor tissue, according to the manufacturer’s protocol. DNA was extracted from tumor tissue using G-spin™ total DNA extraction kit (iNtRON Biotechnology Co., Sangdaewon-dong, Korea), and its concentration was measured at 260 nm according to Glasel 20 using spectrophotometer (Unicam, Leeds, England).
Real-time polymerase chain reaction for cyclin D1
Total RNA was extracted from tumor tissue using RNA-spin™ total RNA extraction kit (iNtRON Biotechnology Co., Korea) under liquid nitrogen. In total, 1–5 µg of total RNA was used for preparing complementary DNA (cDNA) by TIANScript reverse transcription kit (Tiangen Co., Beijing, China; two-step real-time polymerase chain reaction (RT-PCR) kit). cDNA was used for quantitative PCR with SYBR Green I PCR (iNtRON Biotechnology Co., Korea). The RT-PCR program was 95°C, 30 s (pre-denaturation), then 40 cycles (95°C for 5 s for denaturation and 55°C for 10 s for annealing/extension). The target gene Ct values were normalized to the Ct value of the housekeeping gene (β-actin) and expressed as relative copy number (RCN). Primers were obtained from Biosearch Technologies Co. (Petaluma, CA, USA) and were prepared according to Gu et al., 21 as presented in Table 1. The relative content of the gene amplification product was calculated using the 2−ΔCt method. 22
Primers for the studied genes in qRT-PCR.

qRT-PCR: quantitative real-time polymerase chain reaction.
Histopathology of tumor tissue
Tumor tissues were fixed in 10% formalin for 24 h and transferred to ascending grades of alcohol, cleared in xylene, and then embedded in paraffin to form blocks. 23 Four micrometer sections were prepared using microtome (Leica RM2135, Wetzlar, Germany), deparaffinized, and stained by hematoxylin and eosin (H&E) stain. Tumor tissues were examined for the histopathological features as well as for the count of apoptotic cells using light electric microscope equipped with spot digital camera and computer program MATLAB software (Olympus Electron Microscope; Shinjuku, Tokyo, Japan).
Statistical analysis 24
Analysis of data was performed with Statistical Package for Social Sciences (SPSS) software version 16.0, and values were expressed as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was used for comparison between groups using Fisher’s least-significant differences (LSD) option, and statistical significance was obtained at p < 0.05. 12
Results
Effect on survival rate and tumor volume
The survival rate was 67% in control tumor group and increased to 80% in DOX group, to 93% in MET group, and to 100% in co-treatment group (Figure 1(a)). The tumor volume in control tumor group on the 12th day was 120.2 ± 5.12 mm3 and increased gradually till it reached 1585.5 ± 168.6 mm3 on the 28th day. Treatment of SEC-bearing mice with MET, DOX, or both drugs exhibited a significant decrease in tumor volume (p < 0.05) on the 28th day compared to control tumor group. On the 28th day, the tumor volume was 1274.4 ± 137.4 mm3 in MET group, 1120.9 ± 113.1 mm3 in DOX group, and 899.5 ± 99.3 mm3 in co-treatment group (Figure 1(b)). Regarding TIR, co-treatment with MET and DOX exerted TIR of 46.7% versus 25.8% in MET group and 33.9% in DOX group (Figure 1(c)).

Effect of co-treatment with MET and DOX on mice bearing SEC: (a) survival rate, (b) tumor volume, and (c) tumor inhibition rate.
Effect on tumor P53, NF-κB, and PAMPK levels
Treatment of SEC-bearing mice with MET, DOX, or both drugs resulted in a significant increase (p < 0.001) in P53 content to 592.77 ± 28.5, 863.08 ± 49.1, and 1606.2 ± 130 pg/mL, respectively, compared to control tumor group (109.46 ± 11.6 pg/mL) (Figure 2(a)). In addition, co-treatment with MET and DOX raised P53 content significantly (p < 0.01) compared to either MET or DOX group.

Effect of co-treatment with MET and DOX on mice bearing SEC: (a) serum P53 level, (b) tumor NF-κB content, and (c) tumor PAMPK content.
MET alone or in combination with DOX significantly (p < 0.001) lowered NF-κB content to 8.12 ± 0.6 and 1.07 ± 0.214 ng/g tissue, respectively, compared to control tumor group (10.3 ± 1.06 ng/g tissue). On the other hand, treatment of SEC mice with DOX resulted in a significant increase (p < 0.01) in NF-κB content (11.2 ± 1.24 ng/g tissue) compared to control tumor group (Figure 2(b)). Combined treatment with MET and DOX significantly (p < 0.001) lowered NF-κB content versus mono-treatment groups.
Figure 2(c) shows that treatment of SEC mice with DOX significantly (p < 0.01) reduced the level of PAMPK (0.1857 ± 0.022 ng/g tissue) compared to control tumor group (0.305 ± 0.038 ng/g tissue). On the other hand, MET alone or co-treatment with DOX and MET caused a significant increase (p < 0.001) in the level of PAMPK to reach 2.26 ± 0.096 and 1.76 ± 0.17 ng/g tissue, respectively, compared to control tumor group. In addition, the level of PAMPK was significantly increased in MET group (p < 0.01) and co-treatment group (p < 0.001) compared to DOX group.
Effect on tumor DNA content
Treatment of SEC-bearing mice with MET, DOX, or both drugs resulted in a significant decrease (p < 0.001) in DNA content (53 ± 11, 41 ± 9, and 12 ± 1 pg/g tissue, respectively) compared to control tumor group (132 ± 15 pg/g tissue) (Figure 3). Co-treatment with MET and DOX lowered DNA content significantly versus MET mono-treatment group (p < 0.001) and versus DOX mono-treatment group (p < 0.01).

Effect of co-treatment with MET and DOX on DNA content in SEC-bearing mice.
Effect on tumor cyclin D1 gene expression
Figure 4 shows that treatment of SEC mice with DOX significantly (p < 0.05) increased cyclin D1 gene expression (0.119 ± 0.023 RCN) compared to the control tumor group (0.0975 ± 0.0095 RCN). On the other hand, MET alone or co-treatment with DOX and MET caused a significant decrease (p < 0.01) in cyclin D1 gene expression to reach 0.0776 ± 0.026 and 0.0207 ± 0.0069 RCN, respectively, compared to the control tumor group. Cyclin D1 gene expression was significantly decreased in MET group compared to DOX group (p < 0.01). Combined treatment with MET and DOX significantly decreased (p < 0.001) cyclin D1 gene expression versus mono-treatment groups.
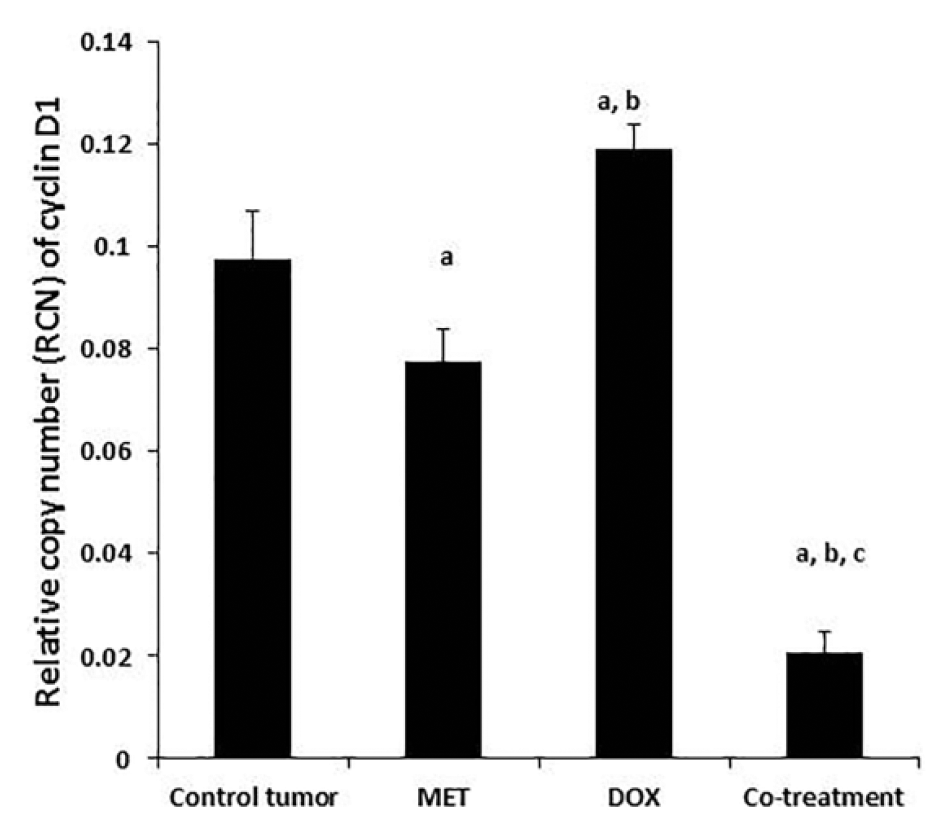
Effect of co-treatment with MET and DOX on cyclin Dl gene expression in mice bearing SEC.
Histopathological results
The histopathological findings are presented in (Figure 5) and (Table 2). Tumor sections from control tumor group showed numerous multinucleated cells, dilated blood vessels, and few apoptotic residual bodies (Figure 5(a)). On the other hand, tumor sections from mice treated with MET showed absence of necrotic cells, normal nuclear/cytoplasmic (N/C) ratio, and significant increase (p < 0.001) in apoptotic cell count up to 1.4-fold of control tumor value (Figure 5(b)). Treatment of SEC mice with DOX resulted in a significant increase (p < 0.001) in apoptotic cell count up to 1.2-fold of control tumor (Figure 5(c)). Co-treatment decreased the number of multinucleated cells, showed normal N/C ratio, and significantly increased (p < 0.001) apoptotic cell count up to 1.6-fold of control tumor (Figure 5(d)). However, the apoptotic cell count in co-treatment group showed a significant increase (p < 0.01) versus DOX mono-treatment group (Table 2).

Photomicrographs of tumor sections from SEC-bearing mice: (a) control tumor group showing a large fibrotic sheet (), groups of multinucleated cells ( ), dilated blood vessels (@), few apoptotic residual bodies (#), and increased apoptosis (**). (b) MET group showing absence of necrotic cells and basophilic cells having vesicular nuclei (
), dilated blood vessels (@), few apoptotic residual bodies (#), and increased apoptosis (**). (b) MET group showing absence of necrotic cells and basophilic cells having vesicular nuclei ( ) and normal nuclear/cytoplasmic (N/C) ratio with increased apoptosis (**). (c) DOX group showing increased apoptosis (**) and (d) co-treatment group showing fewer multinucleated cells (
) and normal nuclear/cytoplasmic (N/C) ratio with increased apoptosis (**). (c) DOX group showing increased apoptosis (**) and (d) co-treatment group showing fewer multinucleated cells ( ), basophilic cells having vesicular nuclei (), and normal (N/C) ratio with increased apoptosis (**).
), basophilic cells having vesicular nuclei (), and normal (N/C) ratio with increased apoptosis (**).
Effect of co-treatment with MET and DOX on apoptotic cell count in mice bearing SEC.

Significant versus control tumor group.
Significant versus DOX group.
Discussion
Cancer is a remarkably complex and heterogeneous collection of diseases that are characterized by uncontrolled cellular growth, local tissue invasion, and distant metastases. 25 Ehrlich carcinoma is a transplantable model for breast cancer. This carcinoma is sensitive to chemotherapy since it is an undifferentiated carcinoma and has a rapid growth rate. 26 In this study, implantation of mice with EAC cells resulted in solid tumor development with a gradual increase in tumor volume within the subsequent 2 weeks. Histopathological findings revealed neoplastic changes such as numerous multinucleated cells and formation of new blood vessels in the tumor tissue of control tumor group.
Because DOX is toxic and causes serious side effects in cancer patients, 10 a major challenge is to lower its side effects without decreasing its effectiveness. MET is a widely prescribed oral medication used as first-line therapy for type 2 diabetes. Previous studies suggest that MET may decrease the incidence of cancer and cancer-related mortality in diabetic patients. 27 In this study, we hypothesized that the anticancer effects of MET might increase the effectiveness of DOX in treatment of SEC mice.
Our results demonstrated that treatment of SEC mice with MET alone or in combination with DOX increased the survival rate and reduced the tumor volume of SEC mice. These results were in agreement with several in vivo–based studies which demonstrated that MET can effectively inhibit growth of the colon, 28 pancreatic, 29 and prostate carcinoma cells. 30
The chemotherapeutic effect of MET was associated with a significant reduction in cyclin D1 gene expression by 20.4% compared to control tumor and by 34% compared to DOX group. Our results were in accordance with Lengyel et al. 8 who reported that MET caused inhibition of ovarian cancer growth in mouse models by blocking cell cycle at G0/G1 which was correlated with a decrease in expression of cyclin D1.
Cyclin D1 is frequently deregulated in cancer and is a biomarker of cancer phenotype and disease progression. It is synthesized rapidly during the G1 phase and accumulates in the nucleus, then degraded when the cell enters the S phase. 31 The ability of cyclin D1 to activate the cyclin-dependent kinases (CDKs), CDK4 and CDK6, is responsible for its oncogenic actions and provides an attractive therapeutic target. 32
On the contrary, we found that DOX treatment increased cyclin D1 expression by up to 1.2-fold of control tumor value. These results could be interpreted by the work of Rezaei et al. 33 who reported that DOX induced overexpression of cyclin D1 in human breast cancer T47D cells and in human promyelocytic leukemia cell line HL-60 at the G2/M phase. 34 However, co-treatment with MET and DOX, herein, exhibited a greater inhibition in cyclin D1 gene expression by 78.8% versus control tumor group, 73.3% versus MET group, and 82.6% versus DOX group.
Cellular DNA content is related to the aggressiveness of various neoplasms. Downregulation of cyclin D1 as observed in this work could contribute to reduction in DNA content, where MET and DOX mono-treatments significantly decreased DNA content, with a greater decrease obtained by the combined treatment. Our results were in accordance with Algire et al. 35 who reported that MET caused reduction in DNA content in mouse embryonic fibroblasts and human mammary epithelial cells. In addition, Sinnett-Smith et al. 36 reported that MET reduced DNA synthesis in pancreatic cancer cells and inhibited endometrial cancer cell growth in vivo. 37 Furthermore, Cruet-Hennequart et al. 38 reported that DOX decreased DNA content in cultured human mesenchymal stem cells and in mammary gland carcinoma. 39
The anticancer effect of MET and DOX, as observed in this study, was associated with significant increase in P53 with induction of apoptosis. Histopathological findings showed that treatment of SEC mice with various treatments increased apoptotic cell count which was significant in co-treatment group rather than mono-treatment groups. Upregulation and activation of P53 is an important target for chemotherapeutic drugs. During the process of malignant transformation in human cells, a number of different changes occur in the cells that result in induction and activation of the P53 protein. Once activated, P53 suppresses tumor progression by restricting growth and survival of the tumor cells. Mutations in P53 or the P53 pathway can permit growth and progression of the tumor mass. 40 P53 was reported to be required for MET-induced growth inhibition, senescence, and apoptosis in breast cancer cells.41,42 In addition, Liu et al. 43 reported that DOX caused early activation of P53 in tumor cells that was followed by caspase-3 activation and DNA fragmentation. DOX was reported to induce apoptosis in human colon cancer cells. 44
In this study, MET treatment significantly increased the level of active phosphorylated form of AMPK, while DOX mono-treatment significantly decreased PAMPK level. AMPK is activated when AMP/ATP or adenosine diphosphate (ADP)/ATP ratios rise in the cells due to various physiological stresses, leading to the activation of LKB1. 45 Otherwise, MET can also mimic these stressors and lead to AMPK activation in an LKB1-dependent manner. AMPK phosphorylation inactivates acetyl CoA carboxylase and downregulates mTOR, thus leading to increased apoptosis and autophagy-mediated cell death. 9 The reverse effect of DOX regarding PAMPK level, herein, could be explained based on the work of Gratia et al., 46 who reported that DOX induced nuclear and mitochondrial DNA damage, reactive oxygen species production, and elevated AMP/ATP ratio, resulting in inhibition of AMPK phosphorylation.47,48 Despite the elevated AMP/ATP ratio, the PAMPK level was even diminished by DOX, as DOX could not affect signaling upstream of AMPK including LKB1. 46 DOX-induced inhibition of AMPK is supposed to be responsible for DOX-induced cardiotoxicity. 49 However, this deleterious effect of DOX was overcome in our study by co-treatment with MET where co-treatment group showed a significant increase in PAMPK level in tumor tissue.
It is well established that AMPK stimulates Sirtuin 1 (SIRT1), peroxisome proliferator–activated receptor γ co-activator 1α (PGC-1α), P53, and Forkhead box O (FoxO) factors, all of which can inhibit the NF-κB signaling but with different mechanisms.50,51 Our results revealed that MET and DOX mono-treatments exhibited opposing actions regarding NF-κB where MET significantly decreased it, but DOX significantly increased it, with reference to control tumor group. Previous studies reported that chemotherapeutic topoisomerase inhibitors such as DOX act as potent inducers of the NF-κB pathway. 52 However, although the level of NF-κB was increased by DOX, it may be unable to initiate and activate the subsequent NF-κB–related transcription processes; this is because activated NF-κB by DOX is deficient in phosphorylation and acetylation, so it lacks key post-translational modifications required for transcriptional activation. 53 Since MET in this study increased the active form of AMPK, we could suggest that activation of AMPK-related pathways could contribute to the decreased level of NF-κB by MET mono-treatment. However, it should be noted that the greatest reduction in NF-κB was obtained by the combined treatment with both drugs.
Conclusion
Co-treatment of breast cancer mice with both MET and DOX showed a greater effectiveness than mono-therapy with either drugs. The combined therapy exhibited a marked anti-tumorigenic effect and corrected the proliferation/apoptosis imbalance with improvement of survival rate. This chemopreventive effect was mediated by downregulation of cyclin D1 while increasing the level of the tumor suppressor gene P53. The activity of AMPK and NF-κB was modulated so that they triggered apoptotic pathway rather than proliferative one. The use of MET as an adjuvant therapy with DOX could augment its cytotoxic effects through affecting different molecular targets.
Footnotes
Acknowledgements
The authors gratefully thank Dr Mona A Yehia, Professor of Histochemistry and Cell Biology, Medical Research Institute, Alexandria, for interpreting and conducting the histopathological examination.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from the Nature Science Foundation of Jiangsu Province (BK2012777) and National Nature Science Foundation of China (NSFC, no. 81270979).