
Abstract
Background
Ubrogepant is a successful drug used for the therapy of migraine.
Objectives
The objective of this work was to formulate a novel nanoparticle-based therapy using ubrogepant for optimum therapeutic efficacy by quick and sustained action.
Materials and Methods
The ubrogepant nanoparticles were prepared using the emulsion solvent evaporation method using poly(lactic-co-glycolic) acid (PLGA) as the copolymer. These nanoparticles were tested by intranasal or intravenous route and in the in vitro assays for drug release and in vivo assays for exposure and activity in a migraine model.
Results
The resulting nanoparticles had significant improvement in cellular uptake and reduced efflux in human colon adenocarcinoma cell line (Caco-2) cells when compared to the plain solution of ubrogepant. The in vivo studies indicated a higher concentration of ubrogepant in the brain, though the absorption is slightly delayed when the intranasal route administers the nanoparticles. The nitroglycerin (NTG)-induced migraine model in rats indicated that the nanoparticles by intranasal route showed significant improvement in locomotion, anxiety, pain behavior, and reduced pro-inflammatory cytokines, tumor necrosis factor (TNF)-alpha, and interleukin (IL)-6 in trigeminal nuclei in the brain. A similar concentration of intravenous administration of ubrogepant was used as the comparator.
Conclusion
Intranasal administration of ubrogepant nanoparticles shows potential as an effective drug delivery system for migraine.
Introduction
Migraine, a neurological disease, affects more than 1 billion people across the globe. The symptoms of migraine are episodic, which occur in three phases, previous to and after the primary symptoms of a unilateral, throbbing, or pulsating headache, which lasts from 4 h to 3 days. The occurrence or attack of migraine is also associated with nausea, photophobia, and sensory symptoms such as flashes of light, blind spots, tingling in the arms and legs, nausea, vomiting, and increased sensitivity to light and sound. All these symptoms are highly variable. 1 Migraine treatment includes serotonin receptor 1B/1D agonists, which are called triptans. 2 These agents cause constriction of the blood vessels and hence are not suitable for patients with hypertension or heart disease. 3 Gepants are a new class of migraine drugs that do not possess side effects like triptans, and hence, they represent better safety than triptans with strong efficacy. 4 Gepants are the antagonists of calcitonin gene-related peptide (CGRP), which is a 37-amino acid neuropeptide. 5 CGRP causes dilation of the blood vessels in the cranium and activation of mast cells, promoting neuroinflammation, which ultimately leads to sensitization to pain in the brain. CGRP is expressed in the trigeminovascular system, where it causes the dilation of blood vessels, causes nociceptive transmission, and induces neurogenic inflammation, which leads to increased sensitivity to pain. 6 Many CGRP antagonists show significant effects in the treatment of migraine, which show comparable effects with triptans, without the cardiovascular liability, with ubrogepant as the first oral small molecule CGRP antagonist that is approved for the treatment of migraine. 7
Quick and effective relief of the pain is a vital aspect of successful migraine treatment. It has been evident that intranasal delivery of agents provides a more rapid effect than oral agents. 8 Also, nasal delivery would be useful for patients with nausea or vomiting, and it may induce a direct effect on the trigeminovascular system. 9 Indeed, intranasal delivery of zavegepant has been successful in establishing rapid and safe pain relief in migraine patients. 10
Ubrogepant is a lipophilic compound that is orally administered. It is a potent CGRP antagonist. Though its lipophilic property is useful in the distribution of the drug, ubrogepant being a P-glycoprotein (P-gp) substrate, the permeability in the brain is limited. 11 Since ubrogepant is a safe and effective agent that is orally administered for the treatment of migraine, we prepared a nanoparticle-based formulation of ubrogepant that could be intranasally delivered for the treatment of migraine.
Materials and Methods
Ubrogepant Nanoparticles
Ubrogepant was obtained from Metrochem API Ltd., Hyderabad. Poly(lactic-co-glycolic) acid or PLGA [lactide:glycolide (50:50), molecular weight 30,000–60,000], polyvinyl alcohol (PVA) (Mowiol 4–88), and dialysis membrane (MWCO: 12–14 kDa) were procured from Sigma (MO, USA). All other chemicals and solvents were of analytical grade. Ubrogepant-PLGA nanoparticles were prepared using the w/o/w emulsion solvent evaporation method. 12 The organic phase was prepared by dissolving accurately weighed PLGA and ubrogepant in dichloromethane (2 mL), which was then mixed with the aqueous phase containing PVA in water (2 mL). It was then sonicated and further poured into PVA aqueous phase solution and sonicated at higher power for 60 s. The solvent was evaporated, and the nanosuspension was purified by centrifugation at 10,000 rpm for 30 min at 40°C to get the final nanoparticles. These pellets were washed with water to remove the untrapped drug, and the pellets were redispersed in water. The nanoparticles, thus made, were lyophilized, and their size was determined using a zeta potential analyzer (Malvern), at the scattering angle of 90° at a temperature of 25°C. The size was also confirmed using scanning electron microscopy (SEM) (Zeiss, Oberkochen, Germany). The entrapped compound was measured by high-pressure liquid chromatography (HPLC) (Agilent 1100, CA, USA) using an already-reported method. 13 The drug formulation solution made in dichloromethane was filtered (pore size 0.22 m, Millipore, Darmstadt, Germany) before analysis. For stability, the formulation was stored in a tightly closed vial at two different conditions; the first was at 4°C and controlled humidity of 75% relative humidity (RH), and the second was at 25°C ± 2°C and controlled humidity of 75% RH. After 3 months, samples were recovered, and the stability study was assessed through EE%, particle size, and zeta potential, with visual examination for contamination, sedimentation, or color change. 13
The drug release from the nanoparticle of ubrogepant was tested using the dialysis method. The release of ubrogepant from the PLGA-NP formulation was analyzed at 37°C ± 1°C by the dialysis method. The dialysis sacs were filled with 1 mL of ubrogepant nanoparticle suspension and were immersed in 30 mL of pH 7.4 phosphate-buffered saline (PBS) solution. The sink condition was maintained, and the system was continuously vortexed at 70 rpm. Samples (0.5 mL) were collected at hourly intervals and analyzed with the liquid chromatography–mass spectrometry (LC–MS) method. 14
Cells human colon adenocarcinoma cell line (Caco-2), a Caco-2 (NCCS, Pune, India), cultured in RPMI-1640 medium [with 1% penicillin–streptomycin and 10% (v/v) fetal bovine serum (FBS) at 37°C, 5% humidity atmosphere and 5% CO2] were seeded in the wells (10 6 cells/well density), and the plates were incubated overnight. The cells were morphologically examined, washed, and incubated with 50 µg of the ubrogepant nanoparticles per mL 12 at 37°C for up to 4 h. The cells were washed with PBS and lysed using 1% Triton X-100. With the same procedure, the efflux of ubrogepant was also studied. For this, after the incubation of each sample with cells for 2 h, cells were further incubated with Earle’s Balanced Salt Solution (EBSS) for up to 2 h. Ubrogepant concentration in the cell lysate was measured by the LC–MS method. 14
In Vivo Studies
Male Sprague Dawley rats (200–225 g) were group-housed (n = 3). The Institutional Animal Ethical Committee (IAEC) approved the animal study protocol constituted under the Committee for Control and Supervision of Experiments on Animals (CCSEA), with the IAEC approval no. DMIHER/IAEC/24-25/10. The animals were maintained at a controlled temperature of 25°C ± 2°C and humidity, with free access to chow food and water. Nitroglycerin (NTG) (in propylene glycol, Sigma) was used to prepare the formulation for subcutaneous injection in 0.9% NaCl and administered at the dose of 10 mg/kg; the vehicle contained similar solvents, except NTG. Ubrogepant treatment was given after 5 min of NTG treatment. Ubrogepant (as plain drug solution or nanoparticle suspension) was administered containing 100 µg/kg absolute ubrogepant. The treatment groups were only NTG (disease control), NTG with ubrogepant intravenous solution or ubrogepant nanoparticles intravenous solution, NTG with intranasal ubrogepant solution, or ubrogepant nanoparticles intranasal solution. The normal control group was sham-treated with vehicles. These groups underwent the open field test (10 min duration), and immediately after the open field test, they were injected with formalin (50 µL, subcutaneously) in the upper lip to perform the orofacial formalin test (45 min duration). 15 The intravenous injection was given in the tail vein and the intranasal administration was done with a polyethylene tubing attached to an injector. 16 The volume administered was 50 µL in each nostril or 100 µL by intravenous infusion. The intravenous formulation was made using polyethylene glycol in buffered saline, and the nanoparticles were dissolved in PBS for administration.
The effect of treatment on the locomotion of rats was assessed by an infrared actimeter (Orchid Scientific, India). After acclimatization, the activity was measured for 5 min. In addition to the locomotor activity, the time spent in the center of the arena was measured. These parameters were measured after 1 h of NTG administration. 17 For the orofacial formalin test, the rats were injected with 50 µL of formalin 1.5% (v/v) into the right upper lip. Face rubbing was evaluated by a researcher blind to treatments, counting the seconds the animal spent grooming the injected area with the ipsilateral forepaw or hind paw 0–3 min (Phase I) and 12–45 min (Phase II) after formalin injection. After that, the animals were sacrificed, and their blood, brain, and brainstem tissues were collected. The brainstem was carefully separated to dissect the trigeminal nucleus caudalis (TNC). 18 The tissue was rinsed in sterile 0.9% NaCl cold solution, placed in cryogenic tubes, and kept at −80°C. For the gene expression processing of tumor necrosis factor (TNF)-alpha and interleukin (IL)-6 gene expression, ribonucleic acid (RNA) extraction was done, and all the samples were checked for absorbance ratios (260/280 nm) for protein contamination. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used for normalization. The primers used were as previously reported. 15
Apart from the behavioral and gene expression studies, the pharmacokinetic analysis was done in a separate group of animals, dosed as above. Brain samples were weighed and homogenized in PBS at pH 7.4. The brain homogenates (200 µL) and serum plasma samples were used for the assessment of ubrogepant concentration by the LC–MS method. 14 The brain and blood were sampled at 0.5, 1, 2, 4, 6, and 8 h. The pharmacokinetic parameters were calculated using WinNonlin software 8.4.
Statistical Analysis
All the results were given as Mean ± Standard Deviation (SD). All results were compared using the two-way analysis of variance (ANOVA) or Student’s t-test or a suitable comparative test as indicated in the figures. Differences are considered significant at a level of p < .05.
Results
In the present study, ubrogepant nanoparticles were prepared by oil in water emulsification and solvent evaporation. The most optimized nanoparticle form had the effect of process parameters used in this emulsification solvent evaporation method on particle size, zeta potential, entrapment efficiency (EE), and polydispersity index (PDI) were investigated. Table 1 shows the details of these nanoparticles. The particle size of 183.7 ± 12.9 was found to be optimum for the nanoparticles. The HPLC method was used that contained the concentration range of 1–500 µg/mL for the entrapment efficiency of ubrogepant (Table 1). The stability data (Table 1) indicate no degradation and change in size or other parameters in the formulation, indicating long-term stability.
Characteristics of Nanoparticle Formulation of Ubrogepant.
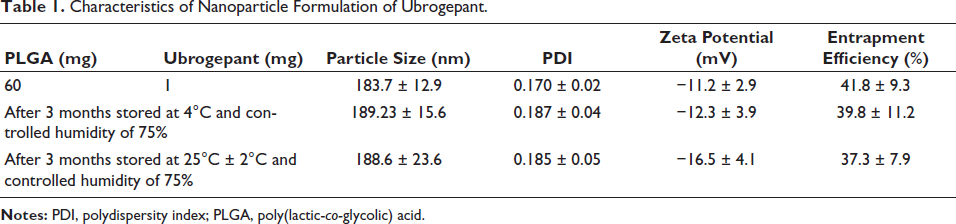
The negative zeta potential of the formulations indicates that the negative charges due to PLGA and PVA could maintain the nanoparticles properly suspended in the formulation. 19 It was also observed that, though the particles tend to aggregate, probe sonication could immediately resuspend the nanoparticles. SEM indicates spherical to oval nanoshapes, with a diameter lower than 200 nm (Figure 1A).
The dialysis method is used to assess the drug release from the nanoparticle formulation. 20 It is observed that the nanoparticles are not released through the dialysis membrane, but the released drug is transported to the receiver compartment. Figure 1B shows the release pattern, indicating that the nanoparticles release the drug in a sustained manner. Since the nanoparticles retained the drug, the diffusion is slow (85% after 24 h), indicating that the drug is properly retained in the formulation.
The absorption and efflux of the nanoparticles by Caco-2 cells showed a time-dependent absorption of the nanoparticles was consistently done by the intestinal cell line (Figure 1C), which was significantly better than the drug in solution in later time points. On the other hand, the efflux of the compound ubrogepant was significantly lower in the case of the drug-loaded nanoparticles when compared to the plain drug solution (Figure 1D). These data indicate that nanoparticles reduce the efflux of ubrogepant from the cells.
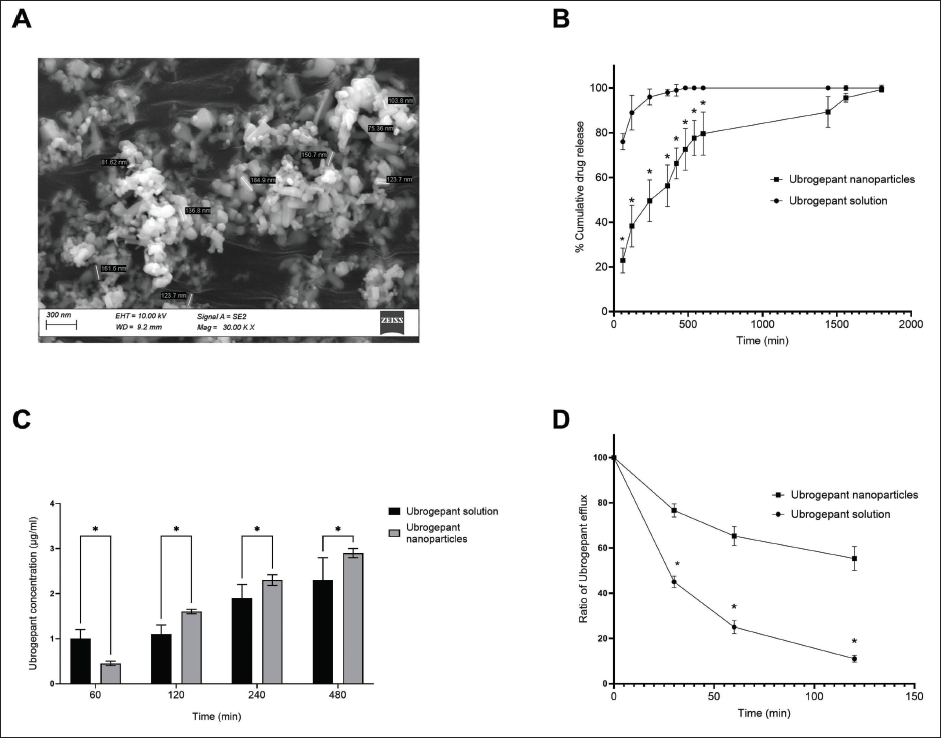
The pharmacokinetic behavior of ubrogepant nanoparticles was assessed along with the ubrogepant drug solution, with doses that contained a similar amount (100 µg/kg) of ubrogepant active compound, and the brain and blood levels of ubrogepant were calculated. The blood levels of ubrogepant were the highest when the intravenous route gave the drug solution.
The pharmacokinetic parameters are shown in Table 2. The intravenous ubrogepant as a free drug solution or nanoparticles demonstrated quick peak formation with Cmax at 30 min, and the drug levels then decreased. In contrast, the Tmax was higher for intranasal administration. Thus, the intranasal administration of ubrogepant showed slower absorption and lesser levels by both the formulations. However, brain concentration of ubrogepant was the highest, as compared to intravenous nanoparticles as well as solution by both routes.
Pharmacokinetic Parameters After Intravenous and Intranasal Administration of Drug Solution and Nanoparticles of Ubrogepant. The Drug was Administered at the Concentration of Absolute Ubrogepant as 100 µg/kg.
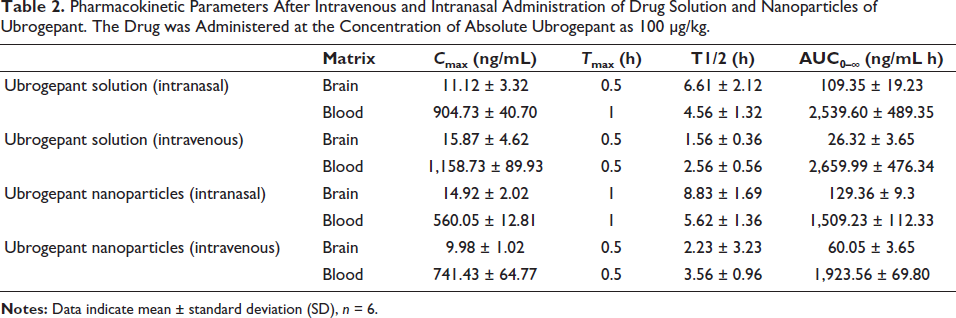
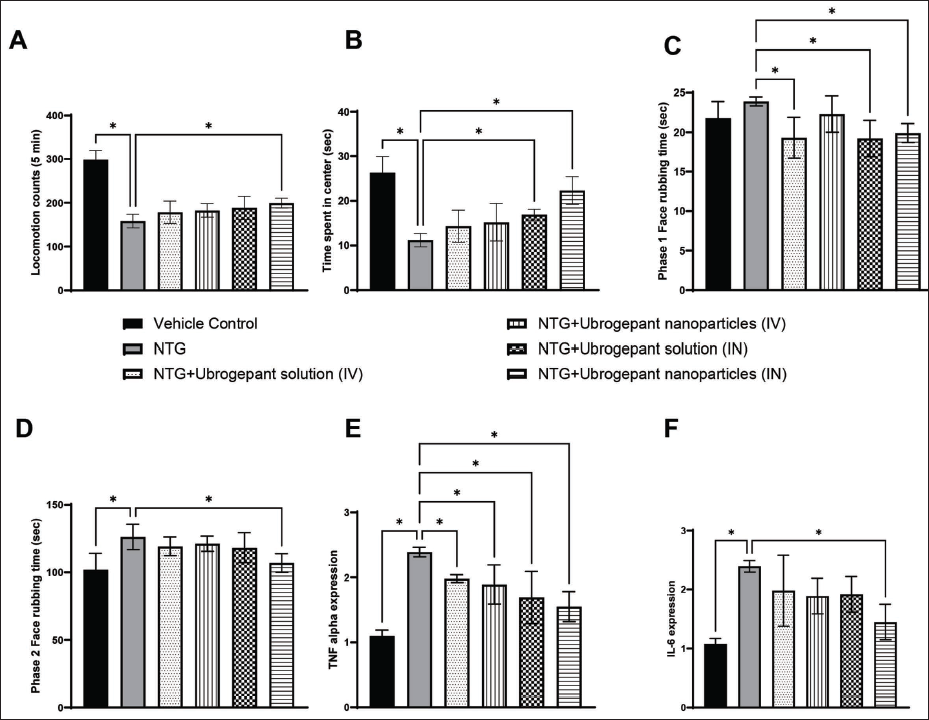
NTG administration significantly reduced the locomotion count and time spent in the central area of the open field. The intranasal administration of ubrogepant nanoparticles clearly reversed the NTG effect in this model. The effect of formalin-induced rubbing effect is indicative of pain, especially in the second phase. This effect was clearly reversed by the intranasal treatment of nanoparticles of ubrogepant. The expression of TNF-alpha and IL-6 was significantly increased by NTG administration, and a consistent and significant reversal of this inflammation was achieved by the intranasal treatment of ubrogepant nanoparticles (Figure 2).
Discussion
Ubrogepant, an oral CGRP receptor antagonist, is used for the treatment of migraine. Migraine is associated with acute pain, which sustains additional symptoms; a rapid onset of action would also have a significant differentiation over the existing therapies. In this regard, the intranasal route of administration could offer a significant advantage for rapid pain relief and sustained effects in the brain. In this work, we demonstrate the profile of the optimized nanoparticle formulation of ubrogepant for the treatment of migraine.
The multiple emulsion using solvent evaporation method indicates that the nanoparticles made had a size below 190 nm, indicating suitability for intranasal administration. 21 The negative zeta potential indicated that though the nanoparticles tend to aggregate, a brief sonification would be effective in resuspending the particles at the time of administration. Coupled with the low polydiversity index and high entrapment efficiency, the nanoparticles indicate a beneficial way of formulating drugs for nasal administration. The release of drug from the nanoparticles as measured during dialysis explains that though the nanoparticles shield the drug release when compared to the solution, the release effect is sustained and enough to cause efficacy.
The cellular uptake assay in Caco-2 cells indicated a significant uptake by the cells, and the efflux of ubrogepant was significantly decreased as compared to the drug solution at 30, 60, and 120-min incubation. It is reported that PLGA nanoparticles are taken up by cells by endocytosis, resulting in higher cellular uptake of the entrapped drug. 22 Studies also indicate that the increased concentration in the Caco-2 cells of the ubrogepant could be due to the drug release from the cytoplasm through changes in surface charges when the nanoparticles with the anionic charge are retained for a longer time in the cytoplasm. 22 Owing to the similar properties of the interaction between the anionically charged nanoparticles and the cellular cytoplasm, the escape of the nanoparticles from the cells by P-gp mediated is also significantly reduced in the case of ubrogepant nanoparticles, which seems to be a mechanism-driven optimization of the drug properties in the formulation. 23 Thus, the nanoparticle formulation of ubrogepant supports significantly improved uptake in the cells as well as sustained retention by inhibition of P-gp.
The nanoparticles slightly delayed the absorption in the brain, which could be due to the retention of nanoparticles in the nasal membrane. However, the slightly delayed absorption is compensated by the higher concentration in the brain (and in Caco-2 cells), with an increased half-life. Though the half-life was higher in both the intravenous and intranasal groups, the actual Cmax and area under the curve (AUC) were significantly higher in the group where nanoparticles of ubrogepant were delivered by the intranasal route. Thus, the intranasal route of administration has better brain targeting compared to intravenous administration of the same nanoparticles. The PLGA polymers used in the current formulation are important in reducing the mucosal clearance that increases the resident time of nanoparticles in the nasal mucosa. 24
The inflammation in the neurons of the trigeminal region and the resulting pain in the cranial area causes migraine-associated pain. 1 The NTG-induced migraine model is a reliable and translatable preclinical tool for studying the efficacy of therapeutic interventions. 25 We have observed that the expression of the inflammatory markers, namely, TNF-alpha and IL-6, were significantly increased by the NTG treatment, and the reduction in the NTG-induced increase in these inflammatory markers was decreased by the intranasal administration of ubrogepant nanoparticles. Consistent with these markers, the intranasal administration of ubrogepant nanoparticles caused a significant reversal of the in vivo effects of NTG, namely, locomotion and formalin-induced pain reflex.
The nanoparticles show enhanced solubility and significant stability, which could enhance drug absorption. The increase in the extent (though not the speed) of absorption is coupled with the enhanced absorption targeting specific tissues or cells, such as the trigeminal nerve, which is involved in migraine pathogenesis. This targeted delivery has enhanced the concentration of ubrogepant at the site of action, enhancing its efficacy. Further, the nanoparticle formulation appears to have provided a controlled release of ubrogepant, ensuring a sustained and consistent therapeutic effect. This can help maintain effective drug levels over a longer period at the site of action. It is also possible that by delivering ubrogepant directly to the site of action and minimizing its exposure to non-target tissues, nanoparticles can reduce the risk of side effects.
The significant improvement in brain penetration makes ubrogepant a beneficial drug to reduce the frequency of migraine attacks for treatment as well as prevention. It is also noted that gepants as a class do not cause medication-induced headaches, which may improve patient compliance in the case of intranasal ubrogepant for migraine patients. The direct absorption from the nasal mucosa would be particularly useful for patients who cannot take oral medications due to severe nausea or vomiting. 26
While CGRP/CGRP receptor expression in the trigeminal ganglion is involved in migraine pathophysiology, CGRP and CGRP receptors have been found in other peripheral and central nervous system (CNS) sites involved in pain signaling, including the striatum, amygdala, hypothalamus, thalamus, and brainstem. Reports indicate that better brain availability and improved efficacy of the CGRP antagonists can be used to treat trigeminal neuropathy, mechanical allodynia, temporomandibular disorders, diabetic neuropathy, chemotherapy-induced neuropathy, and fibromyalgia.6, 27
Our data indicate that nanoparticles can improve bioavailability by enhancing poorly permeable or soluble drugs’ absorption, dissolution rate, and solubility. The challenge associated with the efflux and lesser penetration has been overcome by nanoparticle intranasal formulation of ubrogepant, which achieved extended therapeutic concentration.
The common side effects of ubrogepant are nausea and somnolence, and it is recommended that the drug should not be used with strong CYP3A4 inhibitors, as this can lead to increased exposure to the drug and potential toxicity. Ubrogepant poses a potential risk of hepatotoxicity, especially in patients with pre-existing liver conditions; hence, it is recommended to use it with caution in patients with hepatic and renal impairment. 28 These potential side effects could be lower if the dose titration is done for intranasal delivery of the nanoparticle formulation of ubrogepant.
Ubrogepant nanoparticles can also be given by oral route. However, the nasal mucosa causes quick absorption of the drug in the blood and ensures faster relief from migraine. Avoidance of first-pass metabolism, direct delivery to the brain, and reduction in gastrointestinal side effects are the advantages of intranasal delivery.
Conclusion
Ubrogepant by intranasal route increases the delivery of the drug in the CNS, especially around areas of interest for migraine treatment. Though it may not increase the speed of absorption, the increase in brain penetration and sustenance of pharmacokinetics and pharmacodynamic effect clearly demonstrates the potential of this treatment. Though the safety and toxicity studies for such formulation need to be tested, this work supports the further development of nanoparticle formulation of ubrogepant for intranasal use in therapy.
Footnotes
Abbreviations
Caco-2: Human colon adenocarcinoma cell line; CCSEA: Control and supervision of experiments on animals; CGRP: Calcitonin gene-related peptide; EE: Entrapment efficiency; FBS: Fetal bovine serum; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; HPLC: High-pressure liquid chromatography; IAEC: Institutional Animal Ethical Committee; LC–MS: Liquid chromatography–mass spectrometry; NTG: Nitroglycerin; PDI: Polydispersity index; P-gp: P-glycoprotein; PLGA: Poly(lactic-co-glycolic) acid; PVA: Polyvinyl alcohol; RH: Relative humidity; TNC: Trigeminal nucleus caudalis.
Declaration of Conflict of Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The animal studies for the present work were approved by the Institutional Animal Ethics Committee (IAEC) of the institute constituted as per Committee for Control and Supervision of Experiments on Animals (CCSEA) directions, with protocol no. DMIHER/IAEC/24-25/10.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Not applicable.