
Abstract
Background
Staphylococcal scalded skin syndrome (SSSS) is a dermatological condition caused by Staphylococcus aureus, characterized by exfoliative toxin B, and its increasing resistance to conventional antibiotics necessitates the search for new therapeutic options.
Aim
The study aimed to investigate the potential of traditional medicinal compounds (TMCs) as potential pharmaceuticals against SSSS using molecular-level research.
Introduction
Staphylococcus aureus, commonly known as S. aureus, is the main cause of SSSS. These infections are a major concern for public health because they are becoming resistant to antibiotics. There is an urgent need for new medications to effectively fight against this infection. The main objective of this study was to assess the interaction between TMC compounds and S. aureus exfoliative toxins ETA and ETB utilizing computational approaches.
Materials and Methods
The investigation selected TMC compounds based on their potential to combat S. aureus infections. To predict binding affinities with the ETB toxin, molecular docking simulations were performed using AutoDock and AutoDock Vina. In order to evaluate the stability and interaction dynamics with the toxins, the most promising compound was subjected to a 100 ns molecular dynamics simulation. Stability was evaluated through various methods.
Results
Liquiritin showed the highest binding affinity, with a docking score of −7.6 kcal/mol. MD simulation confirmed the complex’s stability, and the binding free energy of −17.76 kcal/mol indicated potent inhibitory activity against S. aureus ETB.
Conclusion
Liquiritin, a TMC, effectively inhibits S. aureus toxins ETB, with promising potential for treating SSSS and antibiotic-resistant infections, requiring further research.
Keywords
Introduction
Staphylococcus aureus is a bacterium that has a spherical shape that is non-motile. Its unique feature is its ability to form clusters, which can range from pairings to short chains or grape-like clusters. S. aureus is a highly pathogenic bacterium that can cause a wide range of illnesses, including pulmonary infections, vesicular skin lesions, mastitis, abscesses, bacteremia, osteomyelitis, and endocarditis. 1 It is responsible for most respiratory tract infections, as well as skin-related infections such as staphylococcal scalded skin syndrome (SSSS), cellulitis, folliculitis, and impetigo. 2 Among the many diseases under consideration, SSSS or Lyell’s or Ritter’s disease (Ritter von Ritterschein disease) is recognized as a potentially dangerous illness that afflicts a substantial number of people across the globe. An epidemiological research study estimates the yearly occurrence of SSSS among children aged 8 and below to be around 7.67 cases per million in the United States and 696.4 cases per million in China. 3 It is most common in children and accounts for 98%–100% of cases in the USA, France, and Ireland. 4 A recent Korean study isolated MRSA from 96.2% of SSSS patients. 5 In addition, the mortality rate of SSSS in both pediatric and adult populations exhibits significant variation, with rates below 5% in children and above 60% in adults. 6 An underlying immunodeficiency or comorbidity may potentially link to this discrepancy in fatality rates. SSSS is caused by various factors, one of the important toxins is exfoliative toxins (ETs), which are serine protease family members. These toxins, similar to chymotrypsin, are synthesized and released by S. aureus during the post-exponential growth phase. ETs can induce systemic effects in humans, leading to a high mortality rate. Studies show that ETs have a higher level of esterase activity to induce skin infections, particularly through the use of Boc-GluOPh. 7 Another study also demonstrated that serine proteases have a notable level of selectivity towards desmosomal cadherins, thereby significantly impacting the outermost layers of the skin. Therefore, ETs are a primary subject of interest in investigating skin-related infections caused by S. aureus. 2 There are two types of toxin exfoliative toxin B (ETB) and exfoliative toxin A (ETA) produced by S. aureus. ETB is responsible for causing SSSS and bullous impetigo infections, while ETA causes pemphigus foliaceus and bullous impetigo disease in humans. ETB primarily focuses on the inhibition of desmoglein-1 (Dsg-1), a protein that is essential for intercellular adhesion among keratinocytes. When Dsg-1 is cleaved by ETB, it results in the separation of keratinocytes in the epidermis. This conclusion suggests that the separation of layers of epidermal tissue can occur, as seen in cases of scalded skin syndrome and bullous impetigo. 8 ETB disrupts the integrity of the epidermal barrier and facilitates its dissemination through the bloodstream, potentially causing non-infected lesions. The lack of effectiveness of medications targeting the sequence of ETB has been a significant inquiry. As of 2023, the Drug Bank does not contain any drugs specifically designed to target ETB. Studies have demonstrated the ineffectiveness of treatment alternatives like penicillinase-resistant synthetic penicillins in treating S. aureus-induced SSSS. 8
In order to address this issue, the present study explores the use of naturally occurring plant chemicals as an alternative therapeutic intervention for treating various diseases. Traditional medicinal components (TMCs) are essential constituents used by ancient people to address various diseases, 9 such as urinary and gastrointestinal infections. This investigation involved the retrieval of a range of TMCs, which were selected based on their current utilization in the treatment of skin infections as documented in prior research studies. The five plants that have been identified in the literature are Honeysuckle (Lonicera japonica), 10 Forsythia (Forsythia suspensa), 11 Sophora flavescens (Sophora flavescens), 12 Chinese skullcap (Scutellaria baicalensis), 11 and Astragalus (Astragalus membranaceus). 13 Traditional Chinese medicine primarily utilizes these plants, and a thorough examination reveals the high prevalence of 20 compounds in them. The preview literature results of GC-MS reveal the presence of these compounds in large quantities across all five plants. This suggests that they play a key role in treating skin infections 14 and have potential antibacterial, 15 anticancer, anti-inflammatory, 16 neuroprotective, and cardiovascular effects. 17 The study aimed to predict a new drug capable of inhibiting ETB activity using TMCs. The compounds were retrieved from the PubChem database for a molecular docking study, and the best-docked complex was subjected to molecular dynamic simulation using CHARMM27s force field and gmx_MMPBSA. 18 Molecular dynamics simulations are used to estimate the dynamic properties of intricate systems, offering advantages over conventional experimental studies.
Materials and Methods
Software Requirements
Molecular Graphics Laboratory (MGL) tool version 1.5.6 (
Selection and Preparation of Macromolecule
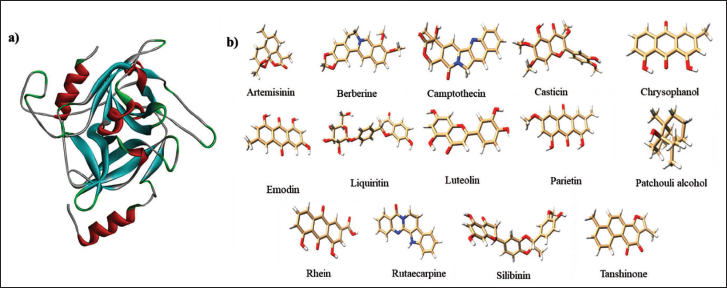
The 3D conformation of ETB was obtained using the RCSB Protein Data Bank (PDB) in .pdb format. The structure of the protein is shown in Figure 1a (1QTF). The Auto Dock 4.2.6 Tool was used to modify the protein’s structure, eliminating heteroatoms and water molecules (exclude water molecules from docking studies, disregarding their impact on binding molecules. Real-time in vivo studies revealed the presence of water molecules at the protein’s active site, increasing interaction between ligand and protein) including hydrogen atoms with polar characteristics, Kollman charges, and AD4-type atoms. The protein file was saved as a protein data bank with partial charge Q and atom type T (pdbqt) format.
(A) 3D Structure of the Exfoliative Toxin B Protein (PDB ID: 1QTF). (B) Specific TMC Bioactive Compounds.
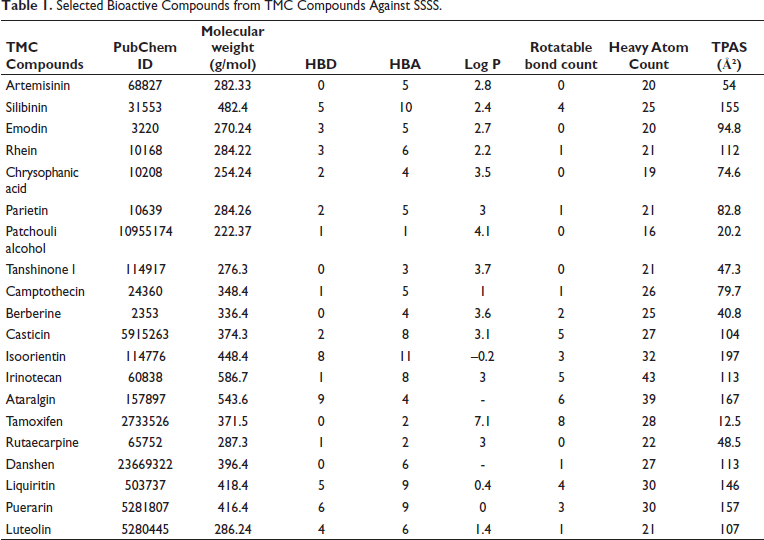
Collection and Preparation of TMC Compounds
The study identified 20 TMC compounds through literature sources and data from the PubChem database. These compounds were visualized using Chimaera software and filtered according to the Lipinski Rule of Five and Ghose Rules 19 . The selected ligands were converted from SDF format to PDBQT format using Open Babel GUI software. Then, using Auto Dock4.2.6 Tools, hydrogen atoms, aromatic carbons, torsion angles, and Gasteiger charges were added to the ligands to get them ready for the docking study. After that, the compounds were extracted and stored in both the PDB file format and the PDBQT format. Table 1 displays the details of the selected TMC compounds, PubChem IDs, and chemical and physical properties.
Selected Bioactive Compounds from TMC Compounds Against SSSS.
Molecular Docking Study
The study focused on investigating the binding interactions between ligands and the 1QTF protein through the use of docking experiments with AutoDock4.2.1 and AutoDock Vina1.1.2 software. For molecular docking, both AutoDock and AutoDock Vina software were used, because AutoDock has strengths like flexibility, customizable parameters, and a robust algorithm, but it is computationally intensive and complex to set up. AutoDock Vina is faster, more user-friendly, and has improved scoring functions and optimization algorithms. Combining both tools provides comprehensive analysis to cross-validate results. The accuracy of scoring functions used for protein–ligand interactions in molecular docking can vary depending on the specific scoring function employed. AutoDock4.2.6 used the Lamarckian Genetic Algorithm (LGA), Monte Carlo simulated annealing, and empirical free energy scoring function techniques to represent the target protein as rigid while presenting the ligand as flexible. 20 The protein and ligand were subjected to AutoDock 4.2.1, generating grid maps using active sites of protein 1QTF (His 65, Asp 114, Ser 186, and Ser 202). 21 The grid box dimensions were 40 × 40 × 40 Å, with a default spacing of 0.375. and center grid box is X = 26.905, Y = 21.883, and Z = 20.504. The docking parameter file (DPF) was configured with a genetic algorithm run of 50 iterations, a population size of 300 individuals, and a maximum evaluation count of 25,000,000. The results were conserved using the Lamarckian Genetic Algorithm and executing the docking parameter file. The same protein was subjected to docking again using Auto Dock Vina software, and the outcomes of the docking studies were compared and analyzed. The docked files were visualized using Discovery Studio and PyMOL tools to find the most favorable binding energies of the complex.
Molecular Dynamic Simulation Study
The Gromacs software program was used for MD simulations using the CHARMM27 force field. The TIP 3-point water model was developed, and protein and the 1QFT-Liquiritin combination were used in simulation studies. Protein topologies were constructed for both ligand-free protein and protein–ligand complex using the CHARMM27 all-atom force field in Gromacs. 22 The SwissParam online web interface was used to create a ligand parameter file compatible with the CHARMM force field. A spatial coordinate file was generated for the ligand topology file. Proteins were immersed in a cubic solvation box to mimic the biological environment, and the system was neutralized using chloride or sodium ions. Energy minimization was performed using the steepest descent minimization algorithm, with the highest recorded force being 10.0 kJ/mol. The system underwent equilibration through NVT and NPT simulations, temperature adjustment to 300 Kelvin, and pressure regulation via the Parrinello-Rahman method. 23 After system stabilization, a MD simulation with a duration of 100 nanoseconds was initiated. The trajectory file was analyzed to compute metrics such as root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration (ROG). Hydrogen bonds formed between the protein–ligand complex were also analyzed. Graphic representations were generated and visualized using the Xmgrace program in the Linux platform.
Calculation of MMGBSA Binding Free Energy
The binding free energy (BFE) of a ligand-protein complex was calculated using the Molecular Mechanics Generalized Born Surface Area (MM-GBSA) method after MD simulation. To determine the BFE, it is required to investigate the ligand’s dynamic nature within the receptor cavity.
24
The BFE was calculated using the last 20 ns of the MD simulation and extracted from the complete 100 ns trajectory using the gmx_MMPBSA program. The process assumed that 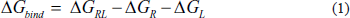
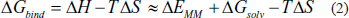
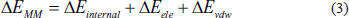
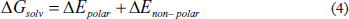
Equation (1) defines the total binding free energy (
Results and Discussion
Selected TMC Compounds and Protein
In this investigation, the TMC substances lying within the molecular weight range of 220–485 g/mol were chosen for further study. Based on this selection criteria, a total of 14 compounds were utilized for the docking investigation (Figure 1b). By analyzing the molecular weights of these compounds, patchouli alcohol exhibited a low molecular weight (222.37 g/mol) whereas silibinin exhibited a high molecular weight (482.4 g/mol). Table 1 displays selected ligands along with their chemical and physical properties.
Molecular Docking of TMC
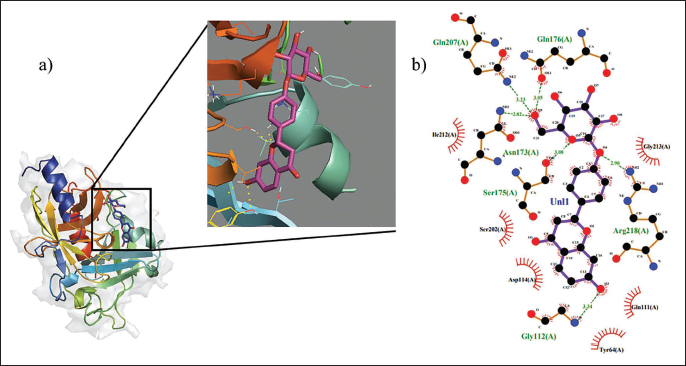
A molecular docking study was conducted to investigate the potential of selected TMC compounds as inhibitors targeting SSSS infection. This study aimed to assess the stability, binding affinity, and interactions of protein–ligand complexes using 1QTF as the target protein. The selected TMC compounds (14 substances) were docked against 1QTF protein using the AutoDock tool and AutoDock Vina software. The docking score validation process entails checking for favorable Van der Waals interaction energies, correct protonation states, and hydrogen bonding patterns. Initially, the process of docking begins by allowing the AutoDock program to interact with the protein. 20 The results of the docked complexes are shown in Table 2 (AutoDock tool and AutoDock Vina results). By comparing the AutoDock tool and AutoDock Vina results, the complex of 1QTF and liquiritin illustrated multiple hydrogen bonds. The AutoDock analysis revealed three hydrogen bonds involving amino acid residues HIS65, SER186, and LEU49, with corresponding bond lengths of 2.1 Å, 2.3 Å, and 2.7 Å. In contrast, the AutoDock Vina results showed five hydrogen bonds with amino acid residues ASN173, SER175, ARG218, GLN112, and GLN207. The AutoDock Vina complex (Figure 2) (1QTF-liquiritin) was chosen for further examination. The study highlights the importance of understanding protein–ligand complexes in drug discovery and development.
Molecular Docking Analysis Conducted Using AutoDock 4.2.1 and AutoDock Vina Software with a Selection of TMC Compounds.
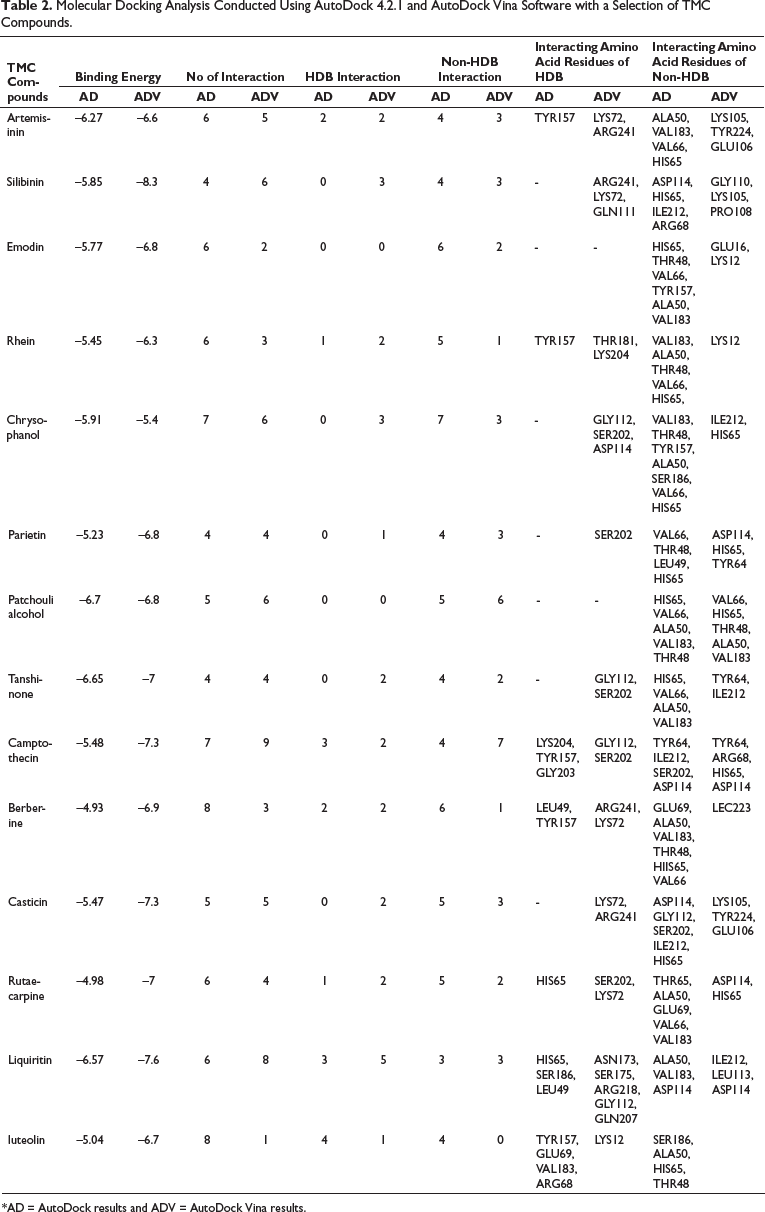
*AD = AutoDock results and ADV = AutoDock Vina results.
(A) 3D Structures View of the Selected Protein Complex (1QTF-liquiritin), with Yellow Dotted Lines Representing the Hydrogen Bonds Formed Between the Receptor and Ligand. (B) 2D Structure View of 1QTF-Liquiritin Complex.
MD Simulation of Protein and Protein–ligand
Molecular dynamics simulations are useful when studying the interactions between receptor–ligand interactions. Following the docking study, the molecular dynamic simulations were performed for 1QTF–liquiritin complex and ligand-free protein in order to evaluate the stability of the small molecule within the binding pocket of the receptor protein. 22 Through MD simulation, RMSD, RMSF, ROG, and H-bond of the protein and complex were determined by utilizing various parameters, such as potential, temperature, pressure, and density. 22
The protein–ligand complex’s conformational stability was assessed using root mean square deviation (RMSD) analysis during MD simulations. Figure 3a illustrates the RMSD of the protein complex (black) and ligand-free protein (red) over a 100 ns duration. The ligand-free protein showed initial RMSD values ranging from 0.08 to 0.125 nm, with significant trajectory fluctuations. The complex showed a consistent backbone RMSD value of about 0.15 nm, confirming its structural integrity. higher RMSD values indicated decreased stability, while smaller values indicated improved stability. This suggests a stable connection between liquiritin and the 1QTF protein.
Outcomes of MD Simulation Including (A) RMSD, (B) RMSF, (C) ROG, and (D) H-bonds. Ligand-free Protein Indicate Red Color, and 1QTF-liquiritin Indicates Black Color Observed Throughout a 100 ns Molecular Dynamics Simulation, and the Binding Free Energy Calculation Last 20 ns are Shown in the (E) representation of various interactions, whereas (F) illustrates the per residue of the 1QTF-liquiritin complex.
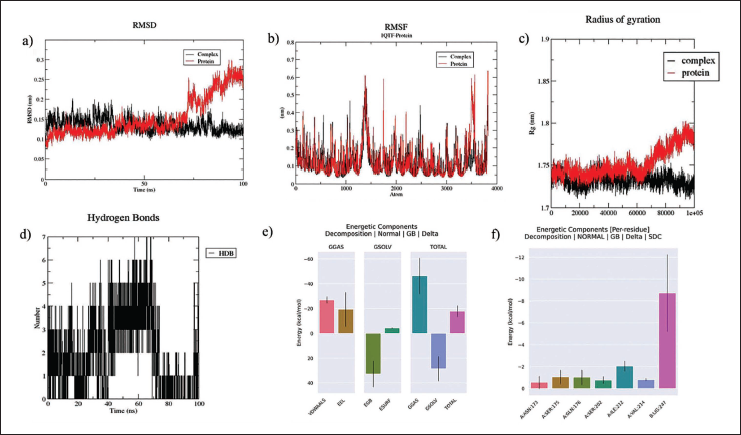
The root mean square fluctuation (RMSF) plot determined the flexibility of residues in the 1QTF protein during and after binding with Liquiritin. Figure 3b illustrates the root mean square fluctuation (RMSF) values of the backbone for both the complex (black) and the ligand-free protein (red), the ligand-free protein had an initial phase value of 0.15 nm, with the highest value recorded at 0.60 nm. The protein–ligand complex had an initial phase value of 0.156 nm, with the greatest degree of fluctuation within the atomic range of 1300–1550 at a wavelength of 0.55 nm. These oscillations led to the destabilization of the protein structure. The RMSF values of the 1QTF-liquiritin complex decreased compared to the protein in the absence of the ligand, suggesting a substantial binding relationship between liquiritin and the 1QTF protein.
The radius of gyration analysis (ROG) was used to assess conformational changes between proteins and ligands, providing information on protein compactness. 27 Figure 3c depicts the protein conformational changes over a 100 ns simulation duration for the 1QTF-liquiritin (black) and ligand-free protein (red). The ligand-free protein had a consistent ROG value of 1.727 for 70 ns, which increased to 1.8 nanometers from 70 ns to 95 ns. The complex ROG value was 1.735 nm for 85 ns, with a minor decline after stable fluctuation. The 1QTF-liquiritin complex exhibited a more stable and compact conformation compared to the protein without ligand.
H-bonds, the strongest non-covalent interactions, are crucial for maintaining the stability of the receptor–ligand complex. The H-bond interaction of the 1QTF–liquiritin complex is shown in Figure 3d. There was a maximum of 6–7 hydrogen bonds along the 100 ns path. Liquiritin showed better binding affinity towards the protein target, highlighting the importance of hydrogen bonds in receptor–ligand complex stability.
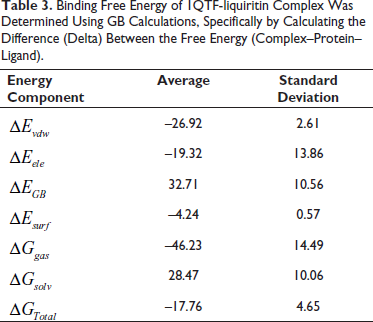
MM-GBSA Calculation and Per-residue Decomposition Analysis
The BFE of the simulated complex was analyzed using the last 20 ns of the trajectory. The complex exhibited a total binding free energy (
Binding Free Energy of 1QTF-liquiritin Complex Was Determined Using GB Calculations, Specifically by Calculating the Difference (Delta) Between the Free Energy (Complex–Protein–Ligand).
Discussion
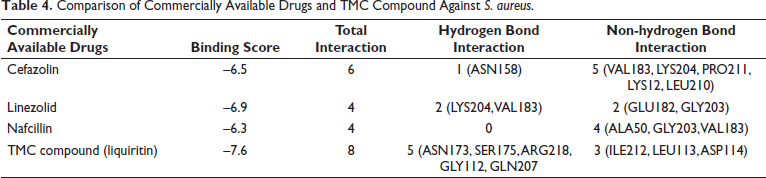
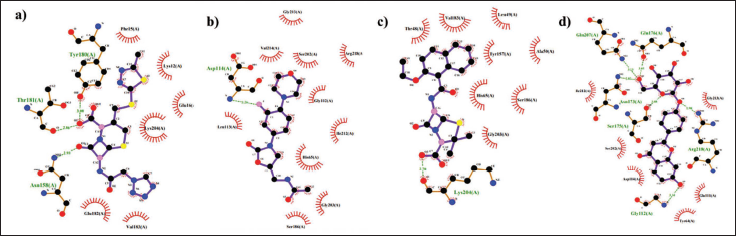
When it comes to SSSS, there are a few commercial inhibitors available on the market; however, they are ineffective in treating this skin condition. It is due to the propensity of pathogenic protein recurrence towards these commercially available drugs as well as the lack of alternative therapies for this skin illness. The study identified one of the promising plant compound Liquiritin, a TMC compound, that exhibited higher binding affinity towards the target protein in comparison to commercially available medications such as cefazolin, linezolid, and nafcillin, which are used for treating S. aureus. The study examined the hydrogen bond and non-hydrogen bond interactions with 1QTF-cefazolin, 1QTF-linezolid, 1QTF-nafcillin, and 1QTF-liquiritin are shown in Figure 4. The detailed comparison between the ligand and commercially available drugs is shown in Table 4. Liquiritin exhibited a higher binding energy of –7.6, surpassing other medications such as cefazolin (–6.5), linezolid (–6.9), and nafcillin (–6.3). The TMC compound exhibited greater inhibitory efficacy compared to commercially accessible medications. The docking was also performed by allowing the protein binding site to be flexible using AutoDock Vina software to find if any conformational changes occurred in the protein binding site during the shift from rigid to flexible protein in the active site. However, there is no conformational change in the protein binding site during flexible active site docking. After undergoing in silico validation, liquiritin is identified to be a potent inhibitor of S. aureus and SSSS infections. Additionally, it has several bioactivities, including anti-inflammatory, antidepressant, anti-Alzheimer’s disease, anticancer, cardiovascular protective, and antitussive characteristics. Additionally, It also has protective properties against skin photodamage, potentially attributed to its capacity to augment the expression of the SIRT3 gene. 29 Therefore, liquiritin exhibits promise as a medicinal agent for safeguarding the skin.
Comparison of Commercially Available Drugs and TMC Compound Against S. aureus.
Comparison of Commercial Drugs and Liquiritin Compounds Against 1QTF. (A) Complexes of 1QTF- cefazolin, (B) complexes of 1QTF- linezolid, (C) complexes of 1QTF- nafcillin, and (D) complexes of TMC compound 1QTF- liquiritin. Figures 4 depicts each complex’s interacting hydrogen bond and non-hydrogen bond interactions.
Conclusion
Staphylococcus aureus infections have posed a persistent threat to human health. This study aimed to conduct an in silico analysis to develop a novel drug targeting ETB, a crucial protein involved in Staphylococcus aureus-induced SSSS infection. The selection of drugs based on the TMC bioactive compounds to target ETB protein (1QTF). Among the selected compounds, Liquiritin was predicted as a new drug based on its better docking score (–7.6kcal/mol) and the higher number of various non-covalent interactions, such as hydrogen bonds, hydrophobic interactions, polar interactions as well as non-polar interactions. Following that, MD simulations were conducted on the docked complex of 1QTF-liquiritin to investigate the ligand conformation. The simulations were carried out for a duration of 100 ns. The results of the simulations indicated that the receptor–ligand complex exhibited favorable structural stability, compactness, and interaction, which were comparable to those observed in commercial inhibitors. In addition to that, the binding free energy analysis provided additional evidence of the strong interaction between liquiritin and the virulent protein. This evidence revealed that liquiritin was a potential inhibitor for targeting ETB. In the future, in vitro and in vivo studies will be carried out in order to assess the efficacy of potential drug development for SSSS infection.
Literature Search Strategy and Selection Criteria
The key search words used were “traditional medicinal compounds,” “ancient plants,” “Staphylococcus aureus,” “Staphylococcal Scalded Skin Syndrome” and “Exfoliative toxin B” in the PubMed and Scopus databases to search for articles written in English only. The articles were selected based on the currently used ancient plants to treat Staphylococcal Scalded Skin Syndrome and skin infections.
Footnotes
Abbreviations
Acknowledgments
The authors would like to thank the management of Vellore Institute of Technology for providing the necessary facilities to carry out this research work.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
Not applicable.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.