
Abstract
Background
The rising incidence of Major Depressive Disorder (MDD), in conjunction with the inadequate effectiveness and resistance observed in existing therapeutic approaches, emphasizes the urgent need to investigate novel therapeutics that provide enhanced efficacy with reduced adverse effects. Abrus precatorius L., an herb that is readily available and frequently utilized in traditional medicine, is frequently prescribed to patients suffering from neurological disorders.
Objectives
The goal of this study is to discover novel active compounds from the A. precatorius plant that can specifically target the GluR3 receptor using in silico techniques, to develop a safe and efficient treatment for MDD.
Materials and Methods
Homology modeling, molecular docking, molecular dynamics simulation, and absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis were employed in this investigation to screen compounds derived from A. precatorius. Their potential to inhibit α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor activity, specifically glutamate receptor 3 (GluR3), which is associated with MDD, was the objective of this study.
Results
Among 31 compounds derived from A. precatorius, 15 successfully cleared the ADMET filter criteria. Subsequently, the best homology model of GluR3 was constructed using Phyre2. This facilitated an investigation into the intramolecular interactions exhibited by the filtered compounds. Notably, the four compounds with the most pronounced binding affinities are identified by their PubChem compound IDs (CIDs): 160511, 44257585, 1983, and 145857. The binding affinity and internal molecular interactions of the top-ranking compound, PubChem CID 160511, were subjected to further validation through MD simulations, affirming its sustained stability in binding.
Conclusion
CIDs: The compound 160511 cleared ADMET analysis with no notable side effects and high binding affinity and stability, making it a potential drug candidate for MDD. To ascertain the precise efficacy of medications, additional in vitro, in vivo, and clinical studies are required.
Introduction
Major Depressive Disorder (MDD) is a common psychological disorder characterized by chronic sadness, loss of interest, and decreased cognitive and physical functioning. Symptoms of MDD include persistently poor mood, despondency, alterations in appetite and sleeping cycles, impaired concentration, and suicidal ideation. Treatment for MDD includes medicine, psychotherapy, and a change of lifestyle (Otte et al., 2016). The World Health Organization (WHO) rated MDD as the third most prevalent illness burden in 2008, and it is anticipated that by 2030, it will surpass all other diseases (Bains & Abdijadid, 2022).
Since new preclinical and clinical studies point to abnormal glutamatergic neurotransmission as a potential therapeutic target, it is imperative to understand the neurobiology of MDD. The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) subtype of ionotropic glutamate receptors is important for synaptic plasticity, which is one of the mechanisms underlying learning and memory (Radchenko et al., 2021). AMPA receptors (AMPARs) are heterotetrametric complexes made up of the subunits GluA1–4. Each of these subunits gives the active receptor unique features, such as trafficking motifs that are connected to synaptic plasticity (Gasbarri & Pompili, 2014; Hollmann & Heinemann, 1994; Shepherd & Huganir, 2007). AMPARs expedite central nervous system excitatory synaptic transmission. Long-term potentiation and depression of synaptic transmission in the hippocampus by these receptors are crucial for synaptic plasticity. AMPARs were initially classified as ionotropic glutamate receptors using selective pharmacological methods. Isolating the genes for the four AMPAR subunits (GluR1-4) has helped us understand their structure, pharmacology, and physiological functions (Jane, 2007). Synaptic activity-related transmission is mediated in adult brains by AMPAR complexes made up of the GluR1 and GluR2 subunits. Synaptic AMPARs are rapidly switched out for GluA1-containing types and kept at a steady level by GluR2- and GluR3-containing receptors in an activity-dependent manner (Banerjee et al., 2016; Henley & Wilkinson, 2016). Despite promising preclinical data linking AMPARs to neuropsychiatric diseases, many AMPAR-targeting drugs have failed to provide therapeutic benefits. This divergence may be due to several factors, but preclinical studies continue to show the therapeutic value of AMPARs in modern antidepressants (Ju et al., 2022).
AMPAR antagonists are being studied as MDD treatments because they may be effective antidepressants. GluR3, an AMPAR subtype, regulates mood (Derkach et al., 2007). More precise and effective depression treatments may result from targeting GluR3 activity. Understanding GluR3’s role in the AMPAR complex helps develop new depressive treatment methods (Sanacora et al., 2008).
Humans and animals need natural products to stay healthy. Their antioxidant, anti-inflammatory, and antiapoptotic properties have been extensively studied. These qualities make the use of phytochemicals good for health and delaying disease onset. Natural compounds have fewer side effects than synthetic drugs, so people seeking alternatives prefer them (Rehman et al., 2019).
The Fabaceae family includes Abrus precatorius L., popularly known as rosary pea, a significant medicinal plant. The biological properties of A. precatorius, including free radical scavenging, renoprotection, neuroprotection, immunomodulatory, anti-inflammatory, anti-lipotoxicity, and antiplatelet aggregation, have been experimentally demonstrated in numerous studies, supporting the plant’s ethnopharmacological uses and other medicinal claims (Boye et al., 2021).
The goal of this study is to discover novel active compounds from the A. precatorius plant that can specifically target the GluR3 receptor using in silico techniques, to develop a safe and efficient treatment for MDD.
Materials and Methods
Selection of Ligand
The phytochemical constituents of A. precatorius were retrieved through literature surveys and online databases, such as Indian Medicinal Plants, Phytochemistry and Therapeutics (IMPPAT) (Mohanraj et al., 2018) and ZINC Database (Sterling & Irwin, 2015). The three-dimensional “.sdf” file of the compounds was retrieved through PubChem (Kim et al., 2021).
Evaluation of In Silico Pharmacokinetics, Metabolism, and Toxicity
This research includes quantitative measurements of drug-like properties such as solubility, absorption, pKa, lipophilicity, blood–brain barrier (BBB), plasma-protein binding (PPB), bioavailability, penetration, permeability, volume of distribution, transporters, skin and ocular irritation, hepatic clearance, and metabolism, among others. The phytochemicals were screened using SMART and the “Filter by Lipinski and Veber Rule” module from Discovery Studio (DS) (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 21.1, San Diego: Dassault Systèmes, 2021). A pharmaceutical molecule’s pharmacokinetic profile, which encompasses absorption, distribution, metabolism, excretion, and toxicity (ADMET), is essential for determining its pharmacodynamic activities. The “ADMET Descriptor” module in DS calculated the ADMET attributes of phytochemicals. Critical to the development of a successful medication is the safety of the chemicals. The “TOPKAT” module in DS was used to assess the toxicity.
Structure Prediction
The GluR3 of AMPAR having accession ID: AAF97857.1 was retrieved from NCBI. The four homology modeling tools (SwissModel, Phyre2, MODWEB, and I-TASSER) were employed for the construction of a three-dimensional structure of AAF97857.1: (a) SwissModel is an automated homology modeling application (Waterhouse et al., 2018; Wu et al., 2003). (b) Phyre2 generates homology models and predicts binding sites (Kelley et al., 2015; Wu et al., 2003). (c) MODWEB is a comparison modeling tool that employs PSI-BLAST to provide automated structural data (Pieper et al., 2006), and (d) I-TASSER is a similar platform that constructs protein structures (Roy et al., 2010). The most potential models from each tool were adopted for further evaluation. ProQ (ProQ—Protein Quality Predictor) and Protein Structure Validation Suite (PSVS) evaluated the best models generated by each application. The “Define Site” module from DS was then applied to this high-quality homology model to identify the binding pocket. The binding pocket of the generated homology model was found to have x, y, and z coordinates of –0.590758, 37.753142, and –3.223064, respectively.
Molecular Docking
The identified compounds from A. precatorius were docked using the “Cdocker” and “LibDock” from DS to explore the binding affinities and modes of binding of the compounds with the protein target (GluR3).
Ligand and Protein Preparation
Ligand structures were processed to assign appropriate atom types, bond orders, and partial charges. The Ligand Partial Charge Method was set to “CFF” in the CDOCKER module to ensure compatibility with the force field used during the docking simulations. Protein structures were prepared by removing water molecules, optimizing hydrogen bond networks, and assigning appropriate charges to polar atoms. These preparations are critical for accurate docking simulations and were carried out in accordance with best practices (Zrieq et al., 2021).
Ligand Optimization with SMART Minimizer
To enhance the accuracy of ligand–protein interactions, the ligands were further optimized using the SMART minimizer from DS (Zrieq et al., 2021). This optimization involved energy minimization of ligand conformations within the protein binding site. The maximum minimization steps were set to 1,000, and the CHARMm force field was applied for this purpose. Ligand optimization helps to alleviate steric clashes and improve the overall quality of the binding poses (Wu et al., 2003).
Molecular Docking with LibDock and CDOCKER
The “LibDock” module, known for its efficiency and robustness, was used for the initial ligand docking simulations. This module considers both polar and apolar interactions and relies on the identification of protein site characteristics known as hot spots. The “Cdocker” module, a molecular dynamics-based approach, was employed to validate and refine the docking results (Zrieq et al., 2021). “Cdocker” utilizes a simulated annealing algorithm to explore ligand binding conformations more comprehensively. Only the optimal docking positions with the lowest binding energies were retained for subsequent analysis (Tasleem et al., 2021, 2023).
Binding Energy Calculation and Intramolecular Interaction Study
The binding energies of the docked ligand–protein complexes were calculated using the “Calculate Binding Energy” module within DS (Zrieq et al., 2021). These energies provide insights into the stability and strength of the ligand–protein interactions. To assess the binding affinity and stability further, we selected the optimum docking positions based on the lowest binding energy for each complex. Additionally, intramolecular interaction analysis was performed to identify key interactions contributing to binding affinity and stability (Tasleem et al., 2021, 2023).
Molecular Dynamics Simulations
Using the Desmond program accessible through Schrodinger Maestro, we ran Molecular Dynamics (Saeed et al., 2021) simulations of the complex to examine GluR3 dynamic activity in simulated physiological settings. The protein–ligand complex was solvated in periodic orthorhombic water-filled TIP3P boxes with a symmetry of 10 10 10 (Jorgensen & Tirado-Rives, 1988). Using OPLS 2005 as a force field, the energy of this solvated device was reduced and its placement was restricted [24]. Additionally, a 100 ns MD simulation was conducted using the NPT ensemble with a 100 ps monitoring interval and 1,000 reading frames at 1 atm pressure and 300 K temperature. Various MD simulation research parameters, such as analysis of ligand binding site, root mean square deviation (RMSD), root mean square fluctuation (RMSF), protein–ligand contacts, and secondary structure element analysis, were investigated to determine the stability, compactness, structural fluctuations, and protein–ligand interactions in a solvated system.
Results
The retrieved A. precatorius phytochemicals were found to have structural diversity, as shown in Figure 1. These 31 phytochemicals with structural variations are highly useful for medicinal purposes.
This study comprises an in silico investigation into A. precatorius phytochemicals that address several molecular mechanisms in both the pharmacokinetics and pharmacodynamics (Silva et al., 2019) stages. In silico pharmacokinetics analysis includes complete drug similarity and ADMET profiling of compounds using various computational tools. To determine the potency of the compounds, in silico pharmacodynamics studies are conducted, which include molecular docking and intramolecular interactions of the phytochemicals with GluR3.
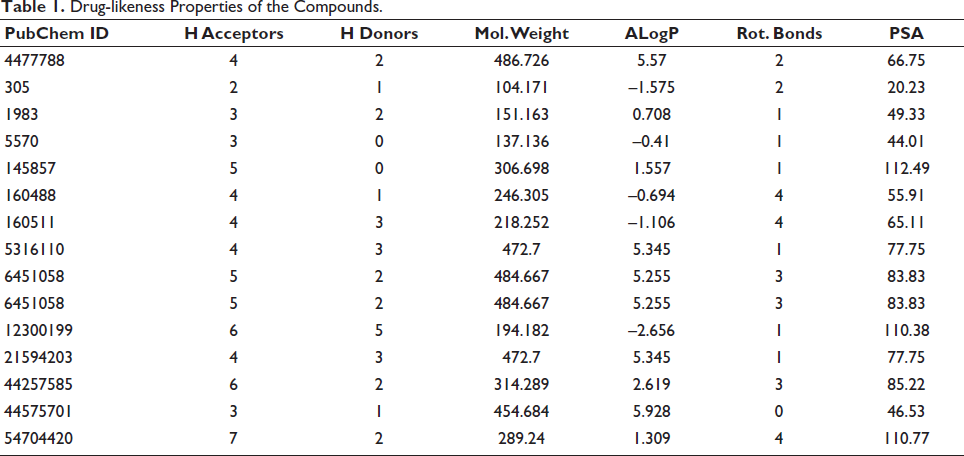
The Lipinski rule identifies molecular properties, such as ADME, required for a drug’s pharmacokinetics in the human body. Each phytochemical’s drug-likeness and ADMET characteristics were analyzed, and the drug-likeness filtered compounds (Table 1) were further investigated.
Drug-likeness Properties of the Compounds.
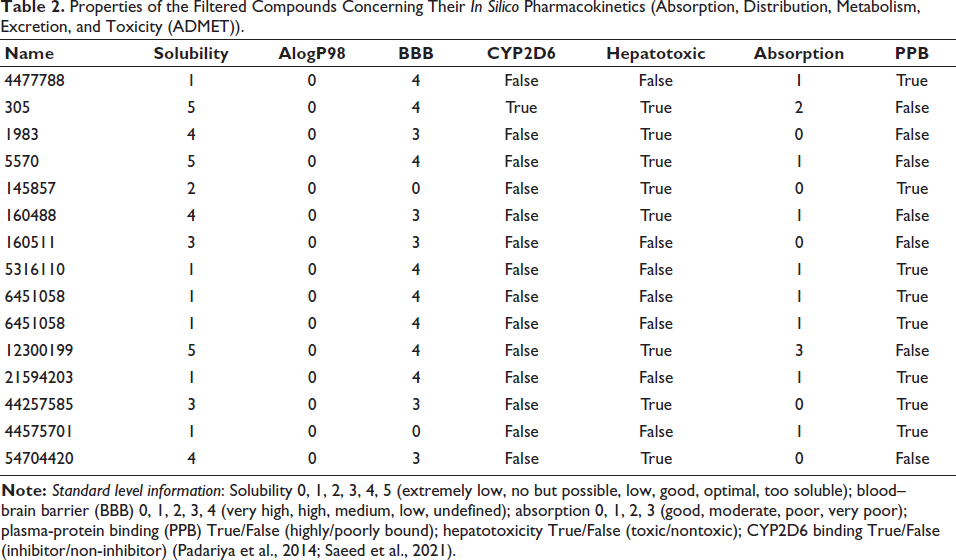
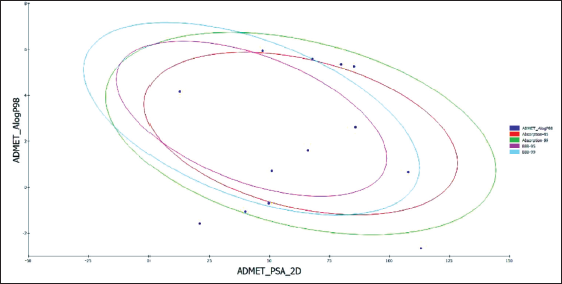
Based on 95% and 99% confidence ellipses, an ADME model was built using the descriptors 2D PSA and AlogP98 to calculate the compounds’ penetration of the BBB and intestinal absorption. Nine phytochemicals with 95% and 99% confidence levels that met the requirements for intestinal absorption and the BBB (Table 2 and Figure 2) were selected for further study. All the filtered compounds were observed to have the best possible solubility and absorption.
Properties of the Filtered Compounds Concerning Their In Silico Pharmacokinetics (Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET)).
Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Plot of the A. precatorius Phytochemicals.
As estimated by TOPKAT and ADMET, while most of the filtered chemicals were found to be non-mutagenic; however, a subset of them were found to be carcinogenic. Few compounds, when taken long-term or in high dosages, cause developmental or reproductive toxicity in addition to mild skin and ocular irritation (Table S1).
Homology Modeling and Best Model Selection
Using PSI-BLAST to search for comparable sequences in the PDB database, it was determined that the AAF97857.1 protein sequence is the most similar to GluR3 from Rattus norvegicus (PDB ID: 6NJM_A) with a similarity of 98.6% and a query coverage of 97%. This sequence was chosen as the template for generating the homology model of AAF97857.1. By employing Phyre2, ModWeb tool, SwissModel, and I-Tasser, four structures of the human GluR3 were generated. Using ProQ and PSVS, the optimal model developed by each tool was validated. The model generated by Phyre2 contains all 232 residues and has the highest LG score, with 91.9% of residues in the most favored region (Table 3).
Validation of the Homology-modeled Structures of the Human GluR3.
Molecular Docking and Interaction Studies
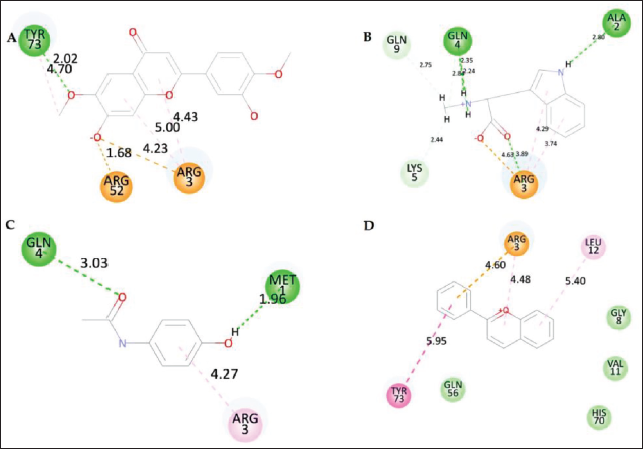
Ten conformations for each ligand were chosen after the CHARMm forcefield and partial charge CFF for ligands were applied. The docked poses with the lowest CDOCKER score and highest LibDock score were selected for the intramolecular interaction analysis, followed by an analysis of the intramolecular interactions of the optimal conformations of the docked poses of the phytochemicals with the target protein. Among the compounds from A. precatorius, ligand 160511 displayed the highest CDOCKER score of 37.28 (Table 4). The interactions have a considerable impact on the compound’s ability to target GluR3 (Table 5 andFigure 3). In compound 160511, salt bridges are crucial for electrostatic interactions (Spassov et al., 2022). The salt bridges in the compound 44257585 with Arg52 and Arg3 and in 160511 with Arg52 and Arg3 are significant as they contribute to the stabilization of the ligand–receptor complex. Conventional hydrogen bonds in compound 44257585 with Arg52 and Tyr73, in compound 160511 with Arg3, Ala2, and Gln4, and in compound 1983 with Gln4 and Met1 enhance binding affinity (Hubbard & Haider, 2010). These bonds facilitate strong molecular interactions, promoting a stable complex. Other hydrophobic interactions, such as Pi–alkyl, Pi–cation, and Pi–Pi–T-shaped interactions play a role in hydrophobic interactions, adding to the overall binding strength and enhancing complex stability (Sing et al., 2022; Tasleem et al., 2021).
2D Interaction of Top Four A. precatorius Phytochemicals. (A) 44257585, (B) 160511, (C) 1983, and (D) 145857.
Docking Score and Binding Energy of the Filtered A. precatorius Compounds.
Intramolecular Interactions Formed between AMPAR and A. precatorius Compounds.
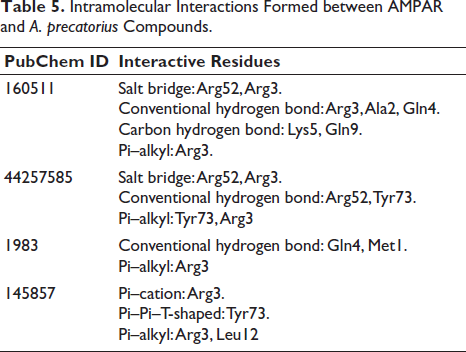
The wide range of interactions observed demonstrates the versatility of these compounds concerning establishing stable complexes with GluR3. The implications of our findings for GluR3-targeting drug development are encouraging. Ligand 160511, specifically, demonstrates substantial intramolecular interactions with critical residues within the binding pocket and exhibits high binding affinities. These top-scoring compounds may serve as lead candidates for further optimization and development into novel GluR3 modulators.
MD Simulation
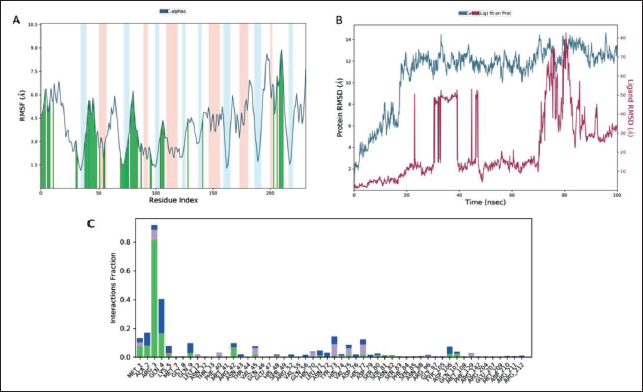
During 100 ns MD simulation of the complex of 160511 with GluR3, a stable complex was formed, which was monitored for RMSD and RMSF to determine the fluctuation in the residues of proteins throughout simulations. Figure 4’s RMSD plot of GluR3 and ligand 160511 provides a broad overview of the structural alterations in proteins that take place during binding. Despite the observed shift in RMSD from 2 to 11 ns, the RMSD plot demonstrated the stability of the GluR3 system. In the simulation analysis, even hydrophobic interactions are retained (Figure 4C).
Bond Formation between Ligand 160511 and GluR3 over 100 ns of MD Simulation (A) Root Mean Square Fluctuation (RMSF) Plot of GluR3 (Blue) and 160511 (Orange), (B) Root Mean Square Deviation (RMSD) Plot of GluR3, and (C) Green, Blue, Gray, and Pink Represent Hydrogen, Water Bridge, Hydrophobic, and Ionic Interactions, Respectively, in the Histogram.
Discussion
In the present study, the three-dimensional structure of A. precatorius retrieved through the IMPPAT database was characterized by unique structural features, which are essential in determining the compounds’ chemical reactivity and possible biological interactions. At first, we come across a range of functional groups, such as the reactive hydroxyl (OH), the adaptable carbonyl (C=O), and the amino (NH2) group. As chemical anchors, these functional groups control reactivity and interactions in chemical reactions as well as possible biological contexts (Yu et al., 1998). Many of these compounds contain aromatic rings, which are well-known for their stability. Compounds with aromatic rings are especially well-suited for a variety of applications, including organic synthesis, materials science, and drug design, due to their intrinsic stability (Polêto et al., 2018). In addition, we investigate compounds that consist of heterocyclic rings. These structures contain atoms like nitrogen, oxygen, or sulfur within the ring framework. They are frequently found in pharmaceuticals and medicinal chemistry, where they contribute to the bioactivity of molecules (Jampilek, 2019). Additionally, certain compounds undergo ionization, which manifests as carboxylate ions (Jorgensen & Tirado-Rives, 1988) or positively charged quaternary ammonium ions (N+). The ionization state of an element has a substantial influence on its solubility, bioavailability, and interactions with biological systems (Fabiano et al., 2020). Halogen atoms, such as chlorine (Clark et al., 2012) and bromine (Br), are added to specific compounds to impact chemical reactivity and increase their ability to dissolve in fats and oils (lipophilicity). The compounds containing halogens are used in various fields, such as medicinal chemistry and agrochemicals (Wilcken et al., 2013). Polyhydroxylated compounds with multiple hydroxyl (OH) groups frequently resemble carbohydrate-like structures. These compounds show promise for possible biological activity and are very important for finding new drugs and studying natural products (Zanardi et al., 2019), as shown in Figure 1. The analysis of these structural properties reveals the complex nature of these compounds, emphasizing their versatility for a wide range of applications, including pharmaceuticals and materials science.
Prioritizing the compounds that have the greatest potential for early ADMET data collection could enhance the screening and testing procedure and lower attrition in the later stages. AlogP98, PSA, PPB, intestinal absorption, aqueous solubility, BBB, hepatotoxicity, and CYP2D6 enzyme inhibition are among the properties examined by the ADMET descriptors module in DS. A linear regression model predicted aqueous solubility at 25°C. Absorption and solubility after oral dosage are indicative of intestinal absorption and drug-likeness. Values for intestinal absorption should be between 0 and 1, indicating good and moderate absorption, respectively, whereas values for aqueous solubility should be between 3 and 4, indicating excellent solubility (Burley et al. 2021; Pu et al., 2022). The BBB score of one of the filtered compounds is 2, whereas the scores of the other compounds are all 3. BBB evaluates how well oral medications reach the central nervous system. A medication with a negative CNS effect would not pass the BBB. The most effective therapeutic substances have BBB values of 3 or 2 (low or medium). Toxicity is evaluated based on a drug’s potential to cause dose-dependent liver injury. The enzyme CYP450 is involved in drug metabolism, and its inhibition is brought by toxic compounds (Ghaffar et al., 2020). 20% of drugs that are processed by the liver are biotransformed by CYP2D6, which makes up 2% of all CYPs. Five CYP isoforms are involved in the metabolism of 80% of drugs tested in clinical trials: 3A4, 2D6, 2C19, 2C9, and 1A2. No substance tested in this study affected the CYP2D6 enzyme, and no liver damage was observed.
Molecular docking is a technique that expedites drug development and reduces the expense of laboratory-based screening by evaluating a compound’s binding affinity, conformation, and poses to a target protein. The possibility of compounds qualifying the drug-likeness and ADMET criteria to bind to the GluR3 protein was assessed. These studies aimed to examine the intramolecular interactions and binding affinities between the protein and the identified chemicals from A. precatorius to rule out false positives. It was observed that hydrogen was generated within the binding pocket by each of the selected compounds. The hydrogen bond formation between the ligand and the protein plays a significant role in binding affinity (Bledsoe et al., 2005). Our docking results demonstrate the successful binding of the selected compounds from A. precatorius within the GluR3 binding pocket. Both the LibDock and CDOCKER algorithms were employed to assess the binding affinity of these compounds, and the results indicate favorable interactions. Among the tested compounds, ligand160511 displayed particularly high CDOCKER scores, indicating strong binding potential. These findings suggest that these compounds have a high likelihood of effectively modulating GluR3 activity. The formation of hydrogen bonds between the compounds and specific residues, such as Arg52, Tyr73, Ala2, Gln4, Lys5, and Gln9, underscores their potential as an effective inhibitor. These interactions contribute to the stability of the ligand–protein complex and are indicative of a strong binding mode (Bulusu & Desiraju, 2020). The observed interactions have a considerable impact on the inhibitors’ ability to target GluR3 effectively. Salt bridges, conventional hydrogen bonds, Pi–alkyl, Pi–cation, and Pi–Pi–T-shaped interactions collectively contribute to the binding affinity and specificity of the compounds (Bosshard et al., 2004). The compound 160511 is an indole alkaloid. The indole alkaloids are nitrogen-rich plant alkaloids identified by their indole ring. Indole alkaloids have been studied for their anticancer, antimicrobial, and central nervous system-modulating properties. These are promising drug discovery candidates due to their unique structure and wide range of biological activities (Bajad et al., 2022). The compounds 44257585 and 145857 are flavonoids. Flavonoids, polyphenolic compounds found in plants, are made up of two aromatic rings linked by a three-carbon chain. Flavonoids are well known for their antioxidant, anti-inflammatory, and anticancer properties. Their distinct pharmacological properties make them promising drug discovery and development candidates (Ullah et al., 2020). Flavonoids have the potential to treat cancer, neurodegeneration, and cardiovascular disorders (Ullah et al., 2020). The compound 1983 is an acetanilide. Acetanilides contain a phenyl ring and an acetamide group (–NHCOCH3). Acetanilides are important in drug discovery due to their wide range of pharmacological activities. Acetanilides have been used in pharmaceuticals, but they are less common in plant products than flavonoids or alkaloids. Acetanilides were investigated for their analgesic, antipyretic, and anti-inflammatory properties (Nasim et al., 2022).
The results of the RMSD analysis demonstrate the stability of the GluR3–160511 complex. The RMSF plot for the GluR3–160511 complex system showed fluctuations in backbone residues in the 180–200 region; this region is significant because it is connected to strong ligand binding. The docking result revealed that the ligand and receptor created many halogen and H-bonds. A dynamic simulation trajectory was then used to further investigate the stability of the ligand. The results analysis revealed that the ligand 160511 has a substantial effect on drug selectivity, metabolism, and adsorption because it maintains a hydrogen bond with the backbone atoms of Met1, Ala2, Arg3, Gln4, Lys5, Gln9, Asp106, and Gln107, according to the results of the dynamic simulation trajectory. The results of the RMSD analysis demonstrate the stability of the GluR3–160511 complex. The RMSF plot for the GluR3–160511 complex system showed fluctuations in backbone residues in the 180–200 region; this region is significant because it is connected to strong ligand binding. The docking result revealed that the ligand and receptor created many halogen and H-bonds. A dynamic simulation trajectory was then used to further investigate the stability of the ligand. The results analysis revealed that the ligand 160511 has a substantial effect on drug selectivity, metabolism, and adsorption because it maintains a hydrogen bond with the backbone atoms of Met1, Ala2, Arg3, Gln4, Lys5, Gln9, Asp106, and Gln107, according to the results of the dynamic simulation trajectory.
During the 100 ns MD simulations, we observed noteworthy conformational changes in the ligand molecules, which have critical implications for their binding to GluR3 receptors. The ligands’ capacity to interact with specific residues in the binding pocket is probably going to be greatly impacted by the conformational changes, which will affect the binding affinity and selectivity (Vogt et al., 2014). Additional research into the ligands’ structural dynamics during binding could provide significant novel understandings of how they bind. MD simulations not only provide insights into ligand dynamics but also shed light on the flexibility and dynamics of the GluR3 receptor itself (Liu et al., 2018). It is important to emphasize that upon ligand binding, particular regions of the protein displayed significant flexibility or conformational changes. To fully comprehend the intricacies of ligand–receptor interactions and how they affect receptor activity, it is crucial to comprehend the dynamics of those proteins. According to our analysis, a crucial step in ligand binding is the creation of hydrogen bonds between the ligands and GluR3 receptors. These hydrogen bonds are essential for preserving the stability of the ligand–protein complex and have a direct impact on the binding affinity. Moreover, the discovery of non-covalent interactions such as halogen bonds highlights the intricacy of ligand–receptor interactions and their roles in binding stability (Chen et al., 2016). One of the key findings from our MD simulations is the extensive network of interactions between the ligand 160511 and specific amino acid residues within the GluR3 receptor binding pocket. These interactions play a pivotal role in determining the stability and binding affinity of the ligand–receptor complex. Specific amino acid residues within the binding pocket played crucial roles in ligand binding. In particular, residues such as Met1, Arg3, Arg13, Gln56, Asn59, His70, Asn72, Tyr73, and Val75 were actively involved in forming hydrogen bonds with the ligands. Understanding the functional significance of these residues is vital for elucidating the molecular mechanisms underlying ligand binding and receptor modulation. The RMSD analysis provides critical insights into the stability of the ligand–receptor complex during the MD simulations. We observed specific trends in the RMSD plots for the GluR3–160511 complex. The RMSD plot for these complexes revealed fluctuations in protein structure within the 2–8 Å range. However, the fluctuations in the RMSD values of the protein fall within 5.5–6.5 Å in the last 10 ns. This indicates a stable equilibrium, suggesting that the ligand effectively interacts with AMPAR without causing significant structural perturbations. The maintenance of equilibrium is a key indicator of the complex’s stability (Aier et al., 2016). While a shift in RMSD was observed from 2 to 11 ns, the RMSD plot displayed equilibrium overall. This finding suggests that the GluR3–160511 complex remains stable throughout the simulation, despite transient structural variations. The stability of this complex is reinforced by the RMSD analysis. The RMSF analysis offers insights into the flexibility and dynamics of protein residues during MD simulations. Notably, the RMSF analysis highlighted specific regions within the protein structure that exhibited greater fluctuations (Dong et al., 2018). The RMSF analysis revealed higher fluctuations in the backbone residues in the region between 180 and 200. This region is particularly interesting as it is associated with potent ligand binding. The increased flexibility of these residues may indicate their active involvement in ligand binding and stabilization. These detailed MD simulation results provide valuable information for drug development targeting GluR3 receptors. The specific amino acid residue interactions, including hydrogen bonds, with Met1, Arg3, Arg13, Gln56, Asn59, His70, Asn72, Tyr73, and Val75, underscore the significance of these residues in ligand binding. The complex’s RMSD plots demonstrate stability, indicating that ligand 160511 forms stable interactions with GluR3 without significantly altering its fundamental structure. This stability shows potential as an indicator for potential drug candidates. Furthermore, the RMSF analysis’s greater flexibility in the region linked to strong ligand binding emphasizes the significance of this region in ligand–protein interactions and proposes possible targets for drug design optimization. A thorough molecular understanding of the interactions between A. precatorius ligands and GluR3 receptors is provided by these MD simulation results. These findings offer critical insights for the rational design and optimization of drug candidates targeting neurological disorders associated with GluR3 receptor dysregulation. It will take more experimental validation and optimization research to utilize these ligands’ therapeutic potential in drug development.
Conclusion
In this study, A. precatorius L. compounds are screened against the GluR3 modeled structure. The compounds that passed the ADMET filter and complied with the drug-likeness were docked in the binding pocket of the GluR3 modeled structure. The compound ID (CID) 160511 was found to possess the highest docking score, which indicated the specific binding mechanism of the selected virtual hit with crucial bond interactions such as hydrogen, van der Waals, and hydrophobic contacts. Molecular dynamics and simulation studies revealed that the compound underwent several conformational changes that allowed it to form stable complexes with the receptor proteins. These findings encourage the potential that these compounds may be acceptable therapeutic candidates for depression. Therefore, additional research and in vitro and in vivo validation are required to develop a phytochemical-based antidepressant that is more potent and effective.
Footnotes
Abbreviations
GluR3: Glutamate receptor subunit 3; ADMET: Absorption, distribution, metabolism, excretion, and toxicity; CID: Compound ID; MDD: Major Depressive Disorder.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
Not Applicable.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research has been funded by the Scientific Research Deanship at the University of Ha’il, Saudi Arabia through project number RG-21 134.
Supplementary Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.