
Abstract
Background
Ischemic stroke is one of the prevalent neurodegenerative disorders; it is generally characterized by sudden abruption of blood flow due to thromboembolism and vascular abnormalities, eventually impairing the supply of oxygen and nutrients to the brain for its metabolic needs. Oxygen-glucose deprived conditions provoke the release of excessive glutamate, which causes excitotoxicity.
Summary
Recent studies suggest that circulatory angiotensin-II (Ang-II) has an imperative role in initiating detrimental events through binding central angiotensin 1 (AT1) receptors. Insufficient energy metabolites and essential ions often lead to oxidative stress during ischemic reperfusion, which leads to the release of proinflammatory mediators such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and cytokines like interleukin-18 (IL-18) and interleukin- 1beta (IL-1β). The transmembrane glutamate transporters, excitatory amino acid transporter-2 (EAAT-2), which express in astroglial cells, have a crucial role in the clearance of glutamate from its releasing site and convert glutamate into glutamine in normal circumstances of brain physiology.
Key Message
During cerebral ischemia, an impairment or dysfunction of EAAT-2 attributes the risk of delayed neuronal cell death. Earlier studies evidencing that angiotensin receptor blockers (ARB) attenuate neuroinflammation by inhibiting the Ang-II/AT1 receptor-mediated inflammatory pathway and that ceftriaxone ameliorates the excitotoxicity-induced neuronal deterioration by enhancing the transcription and expression of EAAT-2 via the nuclear transcriptional factor kappa-B (NF-kB) signaling pathway. The present review will briefly discuss the mechanisms involved in Ang-II/AT1-mediated neuroinflammation, ceftriaxone-induced EAAT-2 expression, and the repurposing hypothesis of the novel combination of ARBs and ceftriaxone for the treatment of cerebral ischemia.
Cerebral Ischemia
Ischemic stroke is one of the prevalent neurodegenerative disorders and is also known as cerebral ischemia. It is generally caused by an abruption of blood flow due to thromboembolism and vascular abnormalities and eventually impairs the supply of oxygen and nutrients to the brain for its metabolic needs. 1 In general, the normal cerebral blood flow (CBF) rate of a healthy adult lies between 50 and 60 mL/100 g/min, which will accomplish the metabolic needs of normal brain physiology. Also, CBF will vary in different regions of the brain. When CBF falls to less than 20 mL/100 g/min, it will commence the activation of the ischemic cascade as a consequence of energy failure, which further diminishes the synaptic activity of neurons, and when it falls to below 10 mL/100 gm/min, irreversible neuronal damage will occur because of oxygen-glucose deprivation (OGD). 2
The pathophysiology of ischemic stroke is a complex, multifactorial phenomenon. However, neuroinflammation contributes a major role in cerebral ischemia; 3 disintegration of the blood–brain barrier (BBB) and activation of microglial cells are the key factors for neuroinflammation and neuronal cell death in cerebral ischemia. 4 OGD in brain cells often instigates subsequent harmful events such as elevated intracellular cytosolic calcium (Ca+2), an impairment of the ionic gradient, which induces persistent cellular depolarization in glial and neuronal cells of the brain. Resultantly, it causes the influx of sodium (Na+) and Ca+2 ions and the efflux of potassium (K+) ions from the neuronal cells and leads to overstimulation of pre-synaptic voltage-dependent Ca+2 channels, thereby increasing the release of excitatory amino acids like glutamate and aspartate in the intrasynaptic space. Elevated glutamate and aspartate concentrations in the synaptic region during energy-depleted conditions activate Ca+2-dependent N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionate acid (AMPA) receptors, which trigger the release of destructive enzymes like protease, lipase, and endonucleases by increasing intracellular Ca+2 levels. The metabolic byproducts of enzymes liberate free radicals and lead to loss of cellular integrity; oxidative stress further activates downstream signaling pathways such as mitogen-activated protein kinases (MAPK) or nuclear factor-κappa B (NF-κB), which release a variety of proinflammatory proteins and cytokines like tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6).5–9
On the other hand, recent studies have shown that accumulation of circulatory angiotensin-II (Ang-II) during cerebral ischemia and other neurodegenerative disorders causes detrimental events like oxidative stress, the release of proinflammatory mediators via overstimulation of angiotensin receptor-1 (AT1) by Ang-II, and microglial activation. It has been proven that blockade of AT1 receptors with angiotensin receptor blockers (ARBs) and angiotensin converting enzyme (ACE) inhibitors attenuates the potential neurodegenerative effects mediating through the Ang-II/AT1 receptor neuroinflammatory pathway.10, 11
Role of Ang-II in Cerebral Ischemia
Generally, the renin-angiotensin-aldosterone system regulates the circulatory system and thereby helps in maintaining the homeostasis of the human body. The enzyme renin will be secreted from the tubules of the kidneys, and angiotensinogen is an α-2 globulin primarily produced by the liver. 12 Other than being the primary source of the enzyme Ang-II, it is often synthesized locally and is also found in various vital organs like the heart, lungs, and brain.13–15 Since the brain angiotensin system (BAS) has a crucial role in the activation of the ischemic cascade, it has been considered a significant aspect of the evolution of different mechanisms involved in cerebral ischemia.
Saavedra and his colleague’s research findings state that Ang-II of the brain has an important function in the regulation of CBF, the hormonal system, innate immunity responses, and drinking behavior. It is also reported for its critical role in several neurodegenerative pathological conditions such as cerebral ischemia, malfunctioning of the BBB, abnormal stress responses, and neuroinflammation as a result of AT1 receptor stimulation triggered by circulatory Ang-II in the brain. Recent studies have revealed that the overexpression of human angiotensinogen and renin augmented the neuronal damage due to elevated Ang-II in the OGD mice model. The expression of ACE-II induces the conversion of Ang-II into Ang (1–7), thereby ameliorating neuronal damage by reducing the binding of Ang-II on AT1 receptors in ischemic mice model.16, 17
The nicotinamide adenine dinucleotide phosphate oxidase and endothelial nitric oxide synthase mediated (NADPH oxidase/eNOS) inflammatory pathway was identified in the ischemic animal model of middle cerebral artery occlusion (MCAo). Astonishingly, the overexpression of ACE-II in transgenic ischemic mice has shown a lesser extent of brain damage as compared with normal ischemic mice, clearly indicating that ACE-II has declined the Ang-II-dependent NADPH/eNOS neuroinflammatory pathway in cerebral ischemia. 18 The Ang-II-mediated neuroinflammation mimics the toll-like receptor (TLR) signal transduction mechanism. 19 The elevated circulatory Ang-II and Ang-II/AT1 receptor overexpression activates microglial cells, thus, causing the release of proinflammatory mediators like TNF-α and IL-1β. 20 Also, Ang-II causes inflammation of vascular endothelial cells through activation of vascular cell adhesion protein-1 (VCAM-1) and NF-kB translocation, 21 and the NF-kB inflammatory pathway is often associated with the expression of low-density lipoprotein receptor-1, which further promotes endothelial dysfunction and atherosclerosis. 22
It has been noticed that the incidence of ischemic stroke increased with angiotensinogen promoter and AT1 receptor gene polymorphism, 23 and experimental evidence has suggested that pressor responses were observed after the cerebral ischemia induction by the MCAo model in Sprague–Dawley rats. The pressor effect may be a factor behind the activation of the pro-inflammatory mediator chemokine, monocyte chemoattractant protein-1 (MCP-1) by Ang-II in the brain, which predominantly regulates the blood pressure in the rostral ventrolateral medulla of the brain stem. 24 These research findings signify that the BAS and circulatory Ang-II have a crucial role in neuronal damage through neuroinflammation. 25
Role of ARBs in Neuroprotection
The enhanced expression of AT1 receptors and their activation through Ang-II up-regulate neuroinflammation during cerebral ischemia. Thus, blocking AT1 receptors with ARBs could protect neurons from neuronal damage. 26 The AT1 receptor antagonists not only regulate the blood pressure but also reduce the level of TNF-α and MCP-1 in the cerebral cortex region of the brain after ischemic induction by the MCAo model in spontaneously hypertensive, stroke-resistant rats. 27
Blockade of AT1 receptors with ARBs exhibited neuroprotection by reducing the expression of proinflammatory cytokines such as interleukin-1β (IL-1β) and vascular endothelial growth factor (VEGF). 28 Recent studies revealed that administration of AT1 blockers has significantly increased neuronal plasticity markers such as postsynaptic density protein-95 (PSD-95) and synaptophysin and shown functional recovery from ischemic stroke. 29 The blockade of central AT1 receptors with a non-hypotensive dose of telmisartan (5 mg/kg) has reduced cerebral ischemic injury via antioxidant and anti-inflammatory mechanisms in a murine model of transient focal ischemia. 30
Glutamate Functions and Clearance
Glutamate is a most potential excitatory amino acid in the mammalian central nervous system (CNS), which plays a critical role in action potential generation. Glutamate oversecretion has been found in several neurodegenerative disorders, especially OGD conditions such as cerebral ischemia. The oversecretion of glutamate leads to excitotoxicity, which initiates detrimental events in neuroinflammation. In order to restore normal brain functions, glutamate will be cleared by the astroglial cells immediately after its release from the presynaptic region. Glutamate clearance is an important event in normal brain physiology; disruption of glutamate clearance leads to excitotoxicity-induced neuronal cell death. In general, glutamate plays a critical role in nerve conduction and signal transduction in normal brain physiology.31–33 The primary function of glutamate is neurotransmission, mediating through ionotropic glutamate receptors such as NMDA and AMPA, 34 and it also acts as a secondary energy resource for neurons in essential times like hypoglycemic conditions. 35
Basically, glutamate clearance is tightly regulated by its release and clearance via the excitatory amino acid transporter (EAAT) system. The EAAT system has an essential responsibility in the maintenance of low glutamate concentrations below the excitotoxic level, where brain functions will be normal. 36 The membrane-bound glutamate transporters generally express in different regions of the brain and other parts of the body organs, such as synaptic endings, oligodendrocytes, astrocytes, and the eye retina. Generally, five different subtypes of EAATs (EAAT-1–EAAT-5) have been investigated to date. 37 The EAAT-1 type of transporte is mostly expressed in the cerebellar regions; EAAT-2 is copiously expressed all over the central nervous system; and EAAT-3 is abundantly expressed in peripheral tissues and the brain. EAAT-4 is typically expressed on GABAergic Purkinje cells, especially in the cerebellum, and the EAAT-5 type of transporter usually expresses on the retina.36–38 Out of the five subtypes, EAAT-2 clears the maximum amount of glutamate from the synaptic region after its release; the remaining transporters are not competent to clear the glutamate below the neurotoxic level, where physiological functions of the brain will be normal. 38
The glutamate clearance through EAAT-2 seems to be an active transportation mechanism (Figure 1). Glutamate molecules are transported into the cytoplasm of the astroglial cell via EAAT-2 transporter proteins through co-transport and counter-transport of Na+ and K+ ions. The entire cycle of glutamate clearance begins with the formation of a complex with a substrate molecule, 3Na+ and 1H+ ions, at the outward-facing conformation of the EAAT protein, and then that complex activates a cascade of inward-facing conformations, eventually releasing substrate, sodium, and proton into the cytoplasm of the cell. After that transporter returns to an outward-facing conformation through the counter-transport of a K+ ion and utilizing 1 adenosine triphosphate (ATP), it becomes ready to transport a new substrate molecule in the synaptic cleft.39–41 An insufficiency or failure in the reuptake of glutamate by the EAAT transporter system has been proposed to be a concern in a wide range of acute neurotoxic conditions like cerebral ischemia, stroke,42, 43 and chronic neurodegenerative disorders such as Alzheimer’s disease, 44 amyotrophic lateral sclerosis, 45 and epilepsy. 46
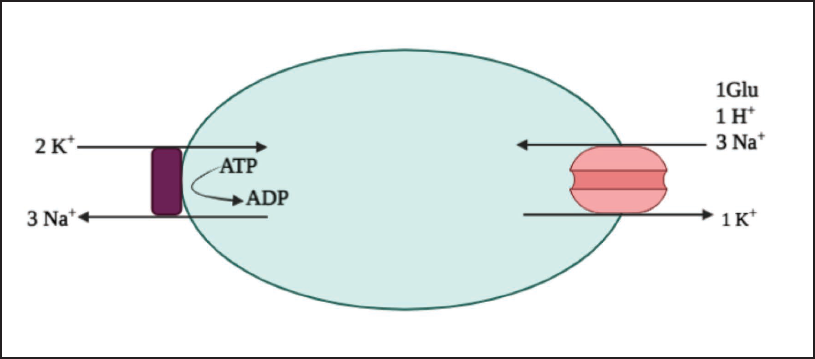
Metabolism of Glutamate
The transported glutamate is metabolized into glutamine in different pathways in the cytoplasm of astroglial cells, and then glutamine enters into the tri-carboxylic acid (TCA) cycle. Glutamate to glutamine conversion generally happens in the presence of an enzyme called glutamine synthetase; it is localized to astroglial cells and oligodendrocytes, but neurons are lacking in this enzyme. 47 Glutamate can also transform into 2-oxoglutarate through an oxidative metabolism via transamination mechanisms or an enzymatic conversion mediating through glutamate dehydrogenase.35, 48, 49 A complete oxidative metabolism of 2-oxoglutarate, when it enters the TCA cycle, generates more than 30 ATPs. Among the 30 ATPs, more than 20 will be utilized to clear the glutamate in each cycle. In addition, metabolic conditions often affect the mechanisms of glutamine conversion and 2-oxoglutarate formation.49, 50 The conversion of glutamine or formation of 2-oxoglutarate depends on the concentration of extracellular glutamate; if the concentration is below 0.2 mM, it is taken up by astrocytes and turned into glutamine by an enzyme called glutamine synthetase; if the extracellular glutamate concentration is above 0.2 mM, it undergoes a transamination reaction, and 2-oxoglutarate will be produced. 49 Meanwhile, various metabolic intermediates such as lactate, alanine, and glutamine will be produced by astrocytes, and they can serve as an energy source during ischemic conditions. 35
EAAT Dysfunction and Oxidative Stress-induced Neurodegeneration
The driving forces required for the clearance of glutamate would be supplied by ATP hydrolysis. An inappropriate sodium/potassium (Na+/K+) ion or functional disability of the Na+/K+ ATPase pump in astroglial cells due to insufficiency in ATP levels unfavorably affects the re-uptake of glutamate.40, 51 The driving forces will be downregulated as a consequence of elevated intracellular sodium and extracellular potassium ion concentrations. The EAAT transporter proteins generally respond to transport the substrate molecules (glutamate) at an invariable membrane potential, which will be maintained by low Na+ levels, high K+ levels in the intracellular environment, and a Na+ ion gradient in the plasma membrane.52, 53 The fluctuations in membrane potential and membrane gradient not only abruptly reverse the reuptake of glutamate by the astrocytes but also reverse the process; as a result, glutamate will leak from the astroglial cells via a Ca
The ischemic insult in the brain leads to OGD. OGD is the foremost cause of excitotoxicity, which generally induces neuronal dysfunction and death. 56 Apart from that, hypoglycemic and hypoxic conditions lead to an alteration in the membrane potential of the neuron and mitochondria, which further diminishes oxidative phosphorylation and thereby impairs glutamate transportation, which in turn elevates glutamate secretion in the intrasynaptic region. Overly secreted glutamate rapidly binds to ionotropic receptors such as NMDA, AMPA, and kinate receptors. The activation of these ionotropic glutamate receptors extensively increases the extracellular Ca+2 levels 57 further, which leads to the activation and release of destructive enzymes like lipase, protease, and endonucleases. In addition, reperfusion of blood brings abundant oxygen to the injured area and the ischemic penumbra region of the brain. The mitochondria, which undergo oxidative stress, could produce oxyradicals in the presence of excess oxygen during ischemic reperfusion. As a result, malfunctioning mitochondria release enzyme complexes and leak electrons into the cytoplasm of the neurons. The leaking enzymes and electrons interact with molecular oxygen and form potent superoxide anions, eventually causing oxidative stress. 58
The oxiradicals like superoxide anions (O2−) combine with nitric oxide (NO) and form (OONO-) peroxynitrite ions, which induce the cytotoxic hydroxyl radicals, leading to the structural disintegration of lipids, nucleic acids, and neuronal proteins,59, 60 as well as oxidative stress and energy failure, which cause DNA fragmentation within mitochondria. Eventually, fragmented DNA will release into the cytosol of the cell; the fragmented DNA segments serve as damage-associated molecular patterns (DAMPs) and trigger the activation of TLR-9, 61 which subsequently activates the NF-kB signaling pathway. Consequently, this cascade promotes the production of various proinflammatory mediators like TNF-α and IL-6.62, 63 In addition, fragmented mitochondrial DNA is involved in the activation of the NLRP3 inflammasome 64 and leads to the release of proinflammatory cytokines such as IL-1β and IL-18, ultimately leading to pyroptotic cell death in neurons. 65
Role of Ceftriaxone in Neuroprotection
Excitotoxicity is one of the main causes of neuronal deterioration in acute and chronic neurodegenerative disorders. 66 Tissue plasminogen activators (tPAs) and thrombolytics are being used as first-line agents for the treatment of stroke, but the efficacy of these drugs is very limited. Thrombolytics and tPAs can recanalize the arterial circulation, although they cannot prevent the neuronal damage caused by excessively secreted glutamate during ischemia. Usually, thrombolytic treatment will be effective upon administration of the drug within 3–4.5 h after the onset of ischemic stroke symptoms. 67 Recent studies have revealed that ceftriaxone also has neuroprotective properties; it upregulates the expression of EAAT-2 on astroglial cells and can also protect the neurons from excitotoxicity-induced neuronal dysfunction in several neurodegenerative diseases.
Third-generation cephalosporin, ceftriaxone (Figure 2) is widely used as a broad-spectrum antibiotic for several bacterial-induced pathological conditions and is also evaluated for neuroprotection in in vitro neuronal cell lines and in vivo animal models. 68 It has been proven that ceftriaxone upregulates the expression of EAAT-2 through NF-kB activation in primary human fetal astrocytes (PHFA). In this process, NF-kB plays a major role in ceftriaxone-mediated EAAT-2 expression, where ceftriaxone directly binds to -272 position (EAAT-2 promoter gene) of astrocyte DNA, which promotes the transcription of EAAT-2 proteins. 69 The activation of NF-kB occurs through the degradation of the cytoplasmic inhibitor, inhibitory kappa-B alpha (IkB-α), from the NF-kB complex, which further results in the liberation of NF-kB, allowing it to translocate the p65 gene and activate the corresponding target genes. These conformational changes lead to an upregulation of EAAT-2 expression on the plasma membrane of astrocytes. Ceftriaxone has significantly protected neurons against global ischemia via upregulating GLT-1 expression in in vivo animal models, 70 and it also restores glutamate transporters and prevents chronic intermittent hypoxia-induced excitotoxic neuronal damage in in vitro P16 organotypic hippocampal slices. 71 Kuo et al.’s research findings stated that administration of ceftriaxone (200 mg/kg) or N-acetylcysteine (150 mg/kg) for 5 days before transient focal cerebral ischemic induction in wister rats developed tolerance to ischemia and significantly depleted the neuronal loss by normalizing the glutamate concentration in the frontal, cortex, and hippocampus of the brain. 72 It has also been reported to attenuate brain injury after subarachnoid hemorrhage by elevating EAAT expression through the PI3K/Akt/NF-kB signaling pathway. 73 In some studies, ceftriaxone has exhibited neuroprotection and improved motor deficits in rodent models of stroke.74, 75
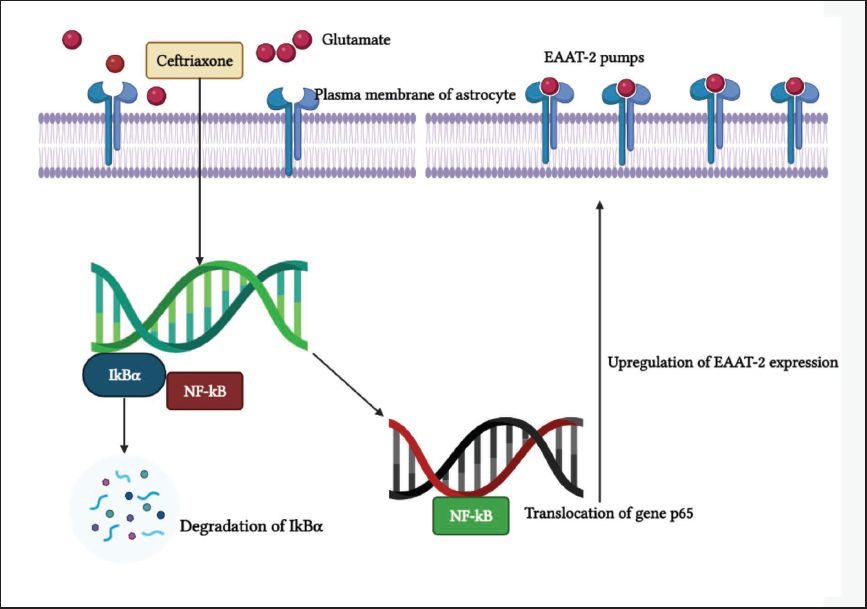
The Current Scenario of Existing Therapeutic Agents and Their Drawbacks
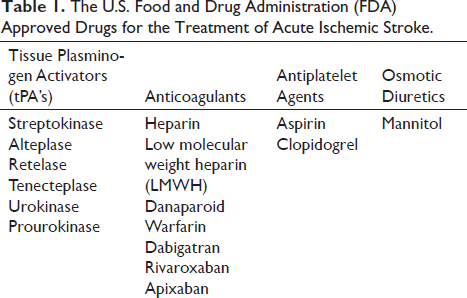
Intravenous thrombolysis with tPAs (Table 1) is a first-line treatment option for the treatment of acute ischemic stroke. tPAs rapidly clear the thrombus and promote reperfusion, thereby helping to protect the neurons from OGD conditions, especially the neurons that are situated in the ischemic penumbra region. Antiplatelet therapy with aspirin and clopidogrel has notably minimized the risk of early recurrence of stroke by inhibiting platelet aggregation; it was proven in clinical trials and is also being used in clinical practice. Anticoagulation therapy is not widely used for the treatment of stroke. The ischemic stroke patients who were treated with anticoagulants such as unfractionated heparin (UFH) and low molecular weight heparin (LMWH) has not shown clinical significance as compared with patients who received antiplatelet therapy. 76 Mannitol, an osmotic diuretic, has been used in clinical practice for the treatment of ischemic and hemorrhagic strokes with normotensive blood pressure. Generally, mannitol lowers the intracranial pressure with its osmatic effect. 77 Apart from the tPAs, antiplatelet agents, anticoagulants, and osmotic diuretics, supportive therapy should be given with antihypertensives, maintenance of appropriate blood glucose levels, and oxygenation for a better outcome.
The U.S. Food and Drug Administration (FDA) Approved Drugs for the Treatment of Acute Ischemic Stroke.
The existing treatment options also have some drawbacks rather than their therapeutic benefits. tPAs are expensive drugs that may not be quickly available in pharmacy stores, and delay in hospitalization often affects the efficacy of thrombolysis. tPAs have to be administered within 3–4.5 h of the onset of a stroke attack. 67 Treatment with streptokinase has been stropped due to high rates of hemorrhage incidence in patients. It should not be used to treat acute ischemic strokes in clinical practice.78–82 The effect of mannitol’s therapeutic benefits on ischemic stroke and cerebral edema is still controversial and unknown; moreover, the existing therapies could not interfere with neuroinflammatory mechanisms. In order to reduce neuronal loss and improve post-ischemic recovery, we need an adjuvant therapy that targets the neuroinflammatory pathways.
Neuroprotective Potential of the Novel Combination ARBs and Ceftriaxone
The AT1 receptor blockers are already being used as antihypertensive agents in clinical practice. Interestingly, telmisartan, valsartan, irbesartan, olmesartan, candesartan, and azilsartan were found to have neuroprotection through their anti-inflammatory and antioxidant mechanisms, as were earlier reports suggesting that the blockade of central AT1 receptors decreased the severity of cerebral ischemic injury in a murine model of transient focal ischemia. 30 Ceftriaxone has been screened as a potential neuroprotective clinical agent that acts as a transcriptional activator for EAAT-2 expression on astroglial cells via the NF-kB signaling pathway. 69 Upregulation of EAAT-2 expression increases glutamate clearance. Thus, it regulates the concentration of glutamate below the excitotoxic level. 70
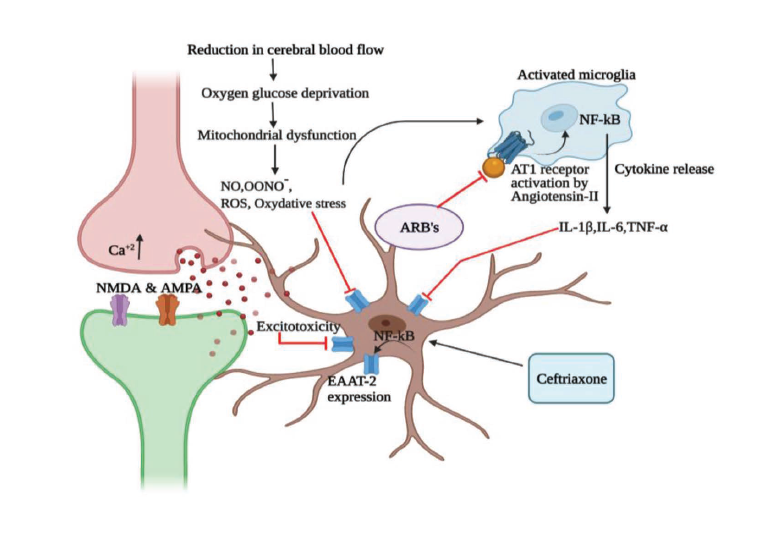
The sudden obstruction in the blood flow impairs oxygen and glucose supply to the brain; oxidative stress will occur as a consequence of ischemic stroke, and eventually, presynaptic neurons release excessive glutamate, which further leads to excitotoxicity. Reactive oxygen species (ROS) and oxidative stress cause the production of cytokines in activated microglial cells. The ROS and cytokine storm often impair the function of EAATs during cerebral ischemia. The AT1 receptors present on the microglial cells will be activated by the circulatory Ang-II; they also have a role in neuroinflammation and cytokine production. ARBs block the AT1 receptors and thus inhibit neuroinflammation and cytokine release from activated microglial cells, which helps to diminish the cytokine-mediated EAAT dysfunction. Ceftriaxone promotes the enhancement of transcription and expression of the EAAT protein on the surface of the astroglial plasma membrane via the NF-kB signaling pathway, which regulates the glutamate concentration below excitotoxicity.
ARBs can attenuate the oxidative stress and malfunctioning of mitochondria, thereby alleviating the risk of excitotoxicity and cytokine storms by inhibiting the Ang-II/AT1 receptor axis (Figure 3); they also exhibit neuroprotection through antioxidant and antineuroinflammatory mechanisms. AT1 blockers can diminish oxidative stress and downregulate the production of cytokines that will be released from activated microglia, but they cannot influence EAAT-2 pumps for rapid clearance of overly secreted glutamate during ischemic conditions. Ceftriaxone has significant potential for glutamate clearance by enhancing EAAT-2 expression on the astrocyte plasma membrane through the NF-kB signaling pathway. Unfortunately, ceftriaxone does not have any role in the regulation of glutamate secretion from the presynaptic cleft region. Both ARBs and ceftriaxone have the capability of neuroprotection in neurodegenerative disorders by showing different mechanisms. The drawbacks of ARBs will be compensated by ceftriaxone, and the drawbacks of ceftriaxone will be fulfilled by ARBs. The novel combination exhibits two different mechanisms on different targets. ARBs arrest the neuroinflammatory pathway in activated microglial cells, which mediates through AT1 receptors and inhibits cytokine storm-induced EAAT-2 dysfunction. Ceftriaxone acts on astrocytes and helps to lessen excitotoxicity through the rapid clearance of glutamate through EAAT-2 pumps. Hence, targeting central AT1 receptors with its antagonists and EAAT-2 transcriptional and expressional enhancement with ceftriaxone could potentiate neuroprotection through antineuroinflammatory mechanisms. Repurposing ARBs and ceftriaxone in a novel combination could be the best treatment option for cerebral ischemia and various neurodegenerative disorders to reduce neuroinflammation and its complications.
The Proposed Neuroprotective Mechanism of the Novel Combination of Angiotensin Receptor Blockers and Ceftriaxone.
Footnotes
Acknowledgements
The authors acknowledge the JSS Academy of Higher Education & Research, Mysuru, India (JSSAHER/REG/RES/URG/54/2011-12/10419) for financial assistance to this work. The authors also thank the DST-FIST, New Delhi, India, for the support towards infrastructure development of Dept. of Pharmacology, JSS College of Pharmacy, Ooty, Tamilnadu, India.
Authors’ Contribution
Gaddam Narasimha Rao and Antony Justin has developed the concept, have done through literature survey, and written the manuscript; Srikanth Jupudi and Antony Justin reviewed and revised for necessary changes in the manuscript, all authors contributed equally.
Statement of Ethics
Not applicable.
Declaration of Conflicting Interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.