
Abstract
Couple screening aims to identify couples with an increased risk of having a child affected with an autosomal recessive or X-linked disorder, in order to facilitate informed reproductive decision making. Both expectant parents should be screened as a single entity, instead of individual testing. Carrier testing was typically performed for a few relatively common recessive disorders associated with significant morbidity, reduced life expectancy and often because of a considerably higher carrier frequency in a specific population for certain diseases. However, new genetic testing technologies enable the expansion of screening to multiple conditions, genes and sequence variants. There are multiple reproductive options for screening couples at risk, particularly when genetic traits are detected in the preconception period.
Keywords
Introduction
The main aim of screening for congenital disorders is to prevent disability with the least harm from side-effects (due to anxiety and unnecessary invasive procedures) and that this is cost-effective. 1 Couple carrier genetic screening attempts to determine, among asymptomatic parents, those with a risk of passing genetic traits to future generations that can lead to autosomal recessive (AR) and X-linked disorders.
There are more than 1300 recessively inherited disorders (autosomal and X-linked), whose symptoms range from the very mild to severe, cumulatively affecting at least 30 in every 10 000 children. 2 This means that approximately 1–2 in 100 couples are at risk of having a child affected with a recessive genetic condition. 3
Standard practice is to offer carrier testing to adults who have a family history of a particular recessive disease. However, the majority of affected children are born to couples with no previous known family history. 4 Preconceptional and prenatal couple screening allow more informed reproductive decision-making, targeted to those who plan to have children.5,6
With AR disorders, only the homozygote person with two allelic pathogenic variants is affected; the heterozygote person with one variant is unaffected and is considered a “carrier” and in many cases is asymptomatic. Since an affected fetus can usually only arise when both partners are carriers, the screening unit is the couple. 7 With X-linked disorders, when a woman is the carrier, she is usually asymptomatic, while male offspring who inherit the pathogenic variant from their mother will develop the disease.
Detailed genetic counseling both prior to and after carrier testing is essential in order for couples to understand the benefits and limitations, potential results and reproductive options. In this review, we will discuss how carrier screening should be applied, and describe different approaches, scope and limitations, pre- and post-test counseling and reproductive options to reduce the risks of having an affected child.
Congenital birth defects
Major congenital anomalies affect 2–3% of births.8,9 Causes of birth defects are many and complex. Single gene defects are responsible for approximately 8% of birth defects.8,9 About 7000 monogenic diseases exist, with different inheritance mechanisms (Table 1). Common AR and X-linked conditions are summarized in Table 2. In high-income countries, single gene defects affect approximately 1% of the population. 9
Single gene disorders: description and global frequency in live births.

MGDb: Modell Global Database of Congenital Disorders.
Christianson A and Modell B. Medical genetics in developing countries. Annu Rev Genomics Hum Genet. 2004; 5: 219–65.
Blencowe H et al. Rare single gene disorders: estimating baseline prevalence and outcomes worldwide. J Community Genet. 2018; 9: 397–406.
Common autosomal recessive and X-linked conditions with their carrier frequency in heterozygosity.

Adapted from Joint SOGC-CCMG committee opinion.
Wilson RD, De Bie I, Armour CM, et al. Joint SOGC-CCMG opinion for reproductive genetic carrier screening: An update for all Canadian providers of maternity and reproductive healthcare in the era of direct-to-consumer testing. J Obstet Gynaecol Can 2016; 38: 742-762.e3.
Preconception genetic counseling involves the analysis of personal and family history in order to determine the risk of birth defects in future pregnancies. In order to identify hereditary diseases, a pedigree should be made, to clarify conditions present in the relatives as well as consanguinity and ethnicity. 10 This allows conditions of AR inheritance and, in some cases, X-linked diseases to be identified. AR diseases are more difficult to identify because usually there is no previous familial history; parents of an affected child are usually healthy carriers.
A non-consanguineous couple has a 1–2% chance of being carriers of a pathogenic variant in the same gene and, as a consequence, being at risk of having offspring affected by recessive diseases.3,11,12
“Carrier screening” vs “couple screening for autosomal recessive disorders”
In the literature, carrier tests are usually referred to as “carrier screening”. A distinction between the two concepts “Carrier screening” and “Couple screening” for AR disorders should be made.
The goal of carrier testing is to identify couples at high risk of having offspring with a serious medical disorder (which occurs when two copies of the recessive gene are inherited). When couples are aware of their risk, they can make an informed decision which might include terminating the pregnancy.
Performing carrier testing in an individual without a partner and no immediate intention of having a child is generally not a satisfactory screening strategy, because:
The object of screening should be the couple. A person who has a carrier status label could feel that they are ‘abnormal’ in some way. The technology evolves year after year, and it is likely that a test that is carried out today will be insufficient in the future, so it will require an update.
If the objective in screening for any autosomal recessive disorder is to identify carriers, almost everyone would be screen positive; moreover, almost all would be false-positives in that they will not have an affected child.
Individual carrier screening is therefore an inefficient and inappropriate approach. Instead, in couple screening, the two expectant parents are a single entity. 7 This should be the screening method of choice because both members of a couple would need to be carriers of the same disorder to be screen positive. This approach directs attention towards the correct yardstick of screening efficacy: the extent to which morbidity and mortality of the medical disorder can be reduced. 13
Approaches to carrier testing for autosomal recessive and X-linked diseases
Carrier testing has traditionally been focused on detecting a select number of genetic diseases, based on patient-reported ethnicity and family history. Limitations of these approaches are imposed by the fact that genetic diseases are not confined within certain populations, and in the setting of increasingly multiethnic populations and more mixed ethnic couples, it is challenging to accurately estimate genetic risk based on ancestry. Options for carrier testing include:
‘Family history-based carrier testing’ is performed when there is an individual in a family who is a carrier or is affected by a genetic disease confirmed by a molecular test Once the pathogenic variant(s) is/are known, the risk of being a carrier is assessed according to the family relationship and a molecular test is performed to detect the pathogenic variant(s) in the other family members. ‘Ethnicity-based carrier testing’ can work well to identify genetic carriers among higher risk populations (e.g., Ashkenazi Jews), but it is dependent on accurate patient-reported ancestry.6,14 Some consider such screening inappropriate.
5
Interestingly enough, those who self-identify with a specific race/ ethnicity may be at odds with ancestry defined genetically, which is of relevance to carrier testing.15,16 ‘Expanded carrier testing’ is a broader approach to carrier testing that has emerged in recent years as a method that takes a pan-ethnic approach to screen for a larger number of genetic diseases.
6
Individuals are screened for hundreds or thousands of selected genetic variants irrespective of reported ethnicity or family history. The main benefit is a better detection rate of carriers.
6
The main disadvantage is that the trained professionals conducting the screening are required to supply additional testing and counselling.
14
Furthermore, finding carriers of rare diseases, with semidominant inheritance, incomplete penetrance or variable expressivity, is common.
Laboratory techniques of carrier tests
Genotyping technology available in the early 1990´s allowed the molecular diagnosis of conditions interrogating a known gene and a few known pathogenic variants. However, in recent years, next-generation sequencing (NGS) technologies have allowed the analysis of a large number of genes. 17 NGS has the advantage of identifying the maximum number of carriers but has the disadvantage of identifying variants of uncertain significance (VUS). A VUS is an alteration in a gene that cannot be categorized with certainty as pathogenic, or as a benign polymorphism. In a carrier testing context, laboratories should not report VUS unless one partner is found to be a carrier of a pathogenic variant in the same gene. 5 Nowadays, screening by NGS is more cost effective than screening for selected mutations in genes. 18
Clinical validity, residual risk and clinical utility of carrier screening
A screening test has clinical validity (good performance) when the detection rate DR (also known as sensitivity) is high, the false-positive rate (FPR) is low and the odds of being affected given a positive result (OAPR) are high. Establishing clinical validity is condition specific. NGS approaches have the potential to increase clinical sensitivity since several thousands of variants are tested. However, sequence variants in several intronic and regulatory genetic regions might remain undetected. Furthermore, certain exonic regions do not yield enough sequencing data to make high-quality genotype detection possible. 19
The OAPR will vary according to the prevalence, penetrance and expressivity of different sequence variants. For prospective carrier couples, the prediction of the phenotype in an affected child will be even more difficult based on the combination of the variants of both parents. Consequently, clinical validity of carrier screening might be uncertain for several variants. 19
‘Residual risk’ arises from the fact that carrier testing cannot identify all heritable conditions because:
- All genes that cause a condition may not be known or may not be examined.
- Causative variants may be in a region not included in the test or may be undetectable by the technology employed.
- Analysis of the gene sequence and its structural variants may be technically difficult.
- Variants may be misclassified with regard to pathogenicity.
An individual's residual risk of being a carrier after having a negative screening test can be calculated as follows: carrier frequency in a population screened x (1 - detection rate (DR) for that population). 17 Therefore, the residual risk of having offspring affected by a given disease after a negative test is calculated by multiplying the residual risk of being a carrier for each member of the couple by the probability of having affected offspring, which would be 25% (1/4). Over time, sequencing data uncovers new pathogenic variants and some are determined to be non-pathogenic as more clinical information becomes available. These changes resulting from new information increase and decrease the detection rate respectively. Furthermore, population carrier frequency varies by ethnicity, and when screening is implemented by simultaneously interrogating multiple variants within multiple genes for rare conditions, the carrier frequency and detection rate may not be known for each condition being screened. 17 The American College of Medical Genetics and Genomics (ACMG) guidelines (2021) state that it is impractical to provide a precise residual risk. Instead, couples should be aware that a negative screening test does not guarantee that they are not a carrier for all conditions, although the risk of being a carrier is greatly reduced. 5
Clinical utility is measured by the fact that couples are informed and may alter reproductive decision-making because of the results. It has been calculated that, for a mixed ethnic couple, the probability of having a pregnancy affected by an autosomal recessive or X-linked condition, detected by an expanded carrier screening test (94 genes), is 1/649. 20 Tier 3 genes proposed by Gregg et al. (2021) 6 were evaluated to determine their potential to identify at-risk couples (Table 3). This showed that the chance of finding an at-risk non-consanguineous couple is, for most ethnicities, less than 1% (except for Africans and Ashkenazi Jews, when it is between 1–2%). 21 In two large studies, one in the United States and one in Europe, a majority (∼60%) took action in response to being identified as an at-risk couple. Reproductive decision-making was more common when couples received results in a preconceptional period (62–77%). The most common decision was to pursue in vitro fertilization (IVF) with a preimplantation genetic test (PGT) (59%), followed by undergoing a diagnostic test during pregnancy (20%), using of a donor gamete (7.7%), and making the decision to adopt (5.1%).22,23
Couples at risk for autosomal recessive disorders expected using the ACMG Tier 3 genes, by ancestry and degree of relatedness.
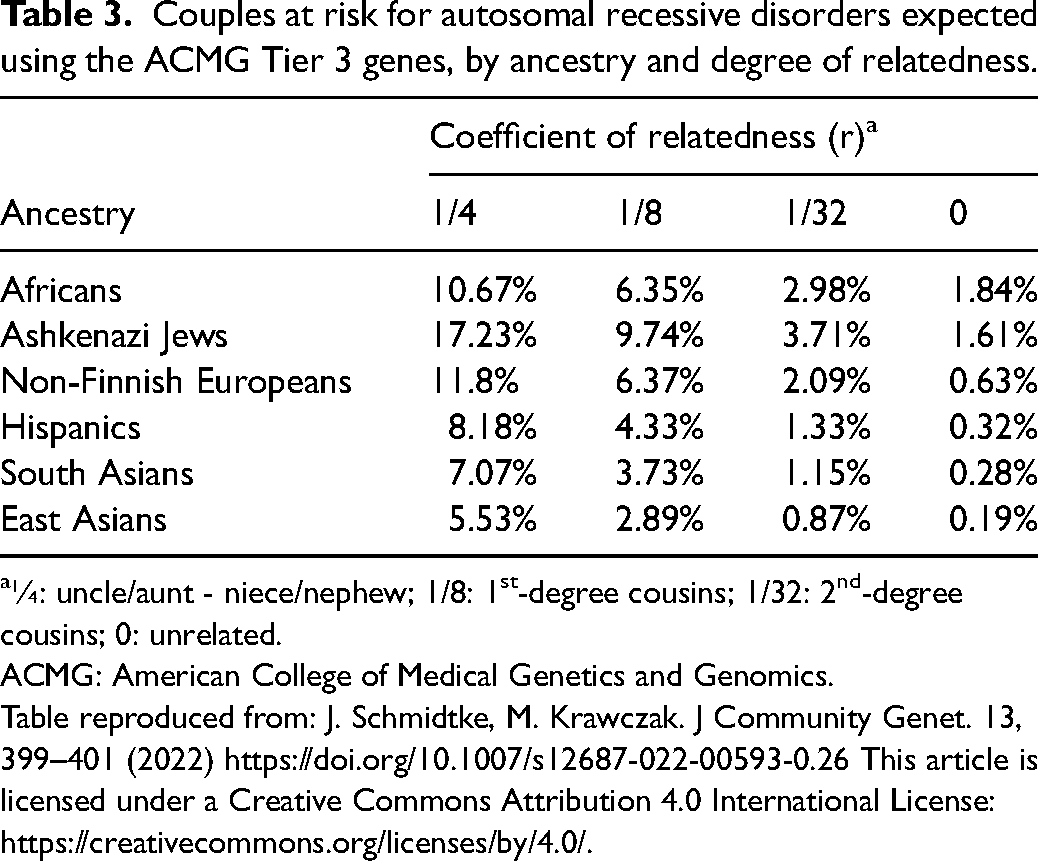
¼: uncle/aunt - niece/nephew; 1/8: 1st-degree cousins; 1/32: 2nd-degree cousins; 0: unrelated.
ACMG: American College of Medical Genetics and Genomics.
Table reproduced from: J. Schmidtke, M. Krawczak. J Community Genet. 13, 399–401 (2022) https://doi.org/10.1007/s12687-022-00593-0.26 This article is licensed under a Creative Commons Attribution 4.0 International License: https://creativecommons.org/licenses/by/4.0/.
Timing strategies for couple screening
Couple screening can be considered before or after conception. Prenatal screening is perhaps more practical since pregnant women are already seeking routine medical attention. However, when couples are screened prior to conception, there is more time to provide detailed pre and post-test genetic counselling, thoroughly address questions, complete follow-up testing and have more reproductive options available.5,6,14,19,24
The procedure in couple screening involves obtaining a test sample, for example a mouth swab, from each partner before any laboratory testing is carried out. One sample is tested and if it does not identify the mutation or mutations being sought the couple is designated screen negative. If the first sample identifies the mutation or mutations being sought the sample from the other partner is tested. If this does not identify the mutation or mutations in question, the couple is designated screen negative. If, however, the mutation or mutations being sought are also identified in the partner's sample, the couple is designated screen positive. This method substantially reduces the false positive rate without a decrease in the detection rate. In couple screening, the couple is screen positive only if both parents (or prospective parents) prove to be carriers of the same disease. 1 Moreover, it allows adequate time for decision-making. An advantage of couple screening is that it tends to avoid complications that arise from non-paternity; the mother can either obtain the second sample from the biological father or simply decline screening. Stepwise screening, by contrast, is effectively carrier screening; each woman is designated screen positive or negative based on the presence of the mutation of locations being sought and the partner approached for a sample only if the mother is positive.
Genetic counselling and topics to discuss with prospective parents
Education and counselling are important in couple screening. Prospective parents should be supported to make informed and autonomous decisions—including the decision not to undergo screening.
17
Topics to cover in pre-test counselling include
5
:
- Education about the conditions being screened for. - Non-consanguineous couples having approximately a 1–2% chance of being at risk of having an affected child with a recessive disorder. - Carrier testing not being a test for all genetic conditions; only a minority are screened for. - Genes and variants may cause a condition that may not be known and may not be examined. - Carrier tests will not identify de novo variants in the offspring. - Carrier tests do not replace newborn screening. - When a reproductive partner has changed, carrier test should be readdressed. - A carrier of a recessive condition will rarely manifest any clinical signs or symptoms of that condition. In some situations, X-linked heterozygous patients will manifest signs and symptoms that are different from the condition seen in offspring. - The concept of residual risk should be addressed: a negative test reduces the chance of having an affected child but does not eliminate the risk. - Variability of manifestations of a genetic condition is typical, even in affected individuals within the same family. - “False positive” results may be due to:
Reduced penetrance of known pathogenic variants; Conflicting variant interpretation among laboratories; Underreporting of outcomes in patients with same variants; Imperfect in silico modeling of variant expression. Additional topics to discuss if an individual happens to be a carrier in step-wise screening
5
: - Offering follow-up screening of the partner with analysis of the same gene that has the pathogenic variant as that identified in the partner. Laboratory testing of the partner should include sequencing of the full gene identified in the carrier patient. - A negative test result in the partner does not eliminate the risk of an affected child. The residual risk cannot be accurately quantified for most conditions, but it is reduced.
Reproductive options
Counselling by an appropriately trained healthcare professional should be part of standard care.
5
Reproductive options that are available to couples include:
No intervention: Having a child naturally and testing after birth to see if the child is affected. Prenatal diagnosis by means of invasive testing: Conceiving naturally and having diagnostic testing during pregnancy to determine if the fetus is affected. This is usually performed with a prenatal invasive test (chorionic villus sampling after 10 gestational weeks or amniocentesis after 15 gestational weeks). DNA is usually extracted to detect pathogenic variants found in parents. This may allow couples to prepare for the birth of a child with a genetic condition, to consider the option of terminating an affected pregnancy, or, in some specific cases, to allow in utero treatment. Prenatal screening by means of cell-free fetal DNA in maternal plasma: Cell-free DNA (cfDNA) can be extracted from a maternal blood sample. Although it is not possible to separate fetal from maternal cfDNA, it has enabled prenatal screening for specific conditions without the associated miscarriage risk that accompanies invasive testing.
25
At present this is mainly used for fetal aneuploidy screening but has the ability to screen for some monogenic conditions from 9 weeks.
25
Confirmatory chorionic villus sampling or amniocentesis is still usually needed. Currently, there are molecular techniques that detect:
Paternally inherited variants for monogenic disorders: in AR disorders, if the paternal pathogenic variant is different to the detected maternal variant, an invasive procedure is required to test for the presence of the maternal pathogenic variant.
25
Maternally inherited variants (X-linked disorders and a definitive diagnosis in AR conditions) are challenging and require dosage-based techniques.25,26 These are currently restricted to a much smaller range of conditions. However, many groups are making advances in this field.
25
Preimplantation genetic testing (PGT). Conceiving the pregnancy by IVF and testing embryos by PGT. Unaffected embryos would then be selected to achieve pregnancy. Using donor sperm, egg or embryo from non-carrying individuals.
Regardless of the timing of a diagnosis, it may be appropriate to refer the couple to see a physician with expertise in the condition that the couple are found to have an increased probability of. The couple should also be offered the opportunity to access community resources and/or a patient support group if available. Affected couples may also consider not having children and adoption.
Discussion
An increasing number of commercial laboratories offer panels for carrier testing for over 100 diseases, though they vary in number and type of diseases included. Implementation of further laboratories in routine healthcare could pose major challenges for healthcare professionals, 19 because carrying out adequate pre and post-test counselling requires knowledge of which methodology is used, which diseases are included, and what the detection rate is.
The American College of Obstetricians and Gynecologists (ACOG) has stated that ethnicity-based, pan-ethnic, and expanded carrier testing are all acceptable strategies. 14 Several scientific societies recommend that couples should be offered carrier screening irrespective of the presence or absence of a family history of a genetic condition, or the ethnicity of the individuals, but none embrace couple screening.5,14,24
A new Clinical Practice Resource released by the ACMG introduced the concept of a tiered structure in defining sets of genes for analysis; they created a set of 113 genes (97 AR with a ≥ 1/200 carrier frequency and 16 X-linked conditions), that should be considered the standard offering to preconception and prenatal patients. 5
In most Western countries there is consensus that the aim of carrier testing should be to enhance reproductive autonomy and enable meaningful reproductive choices, although expanded carrier testing is not much considered in Europe with the exception of assisted reproductive technologies that uses donor gametes. In some other communities with a high burden of severe disease, population-level prevention is regarded as the appropriate aim of screening. 19
While guidelines issued by scientific societies recommend which diseases to include in expanded carrier test panels (Table 4),5,14,27 currently uniform or standardized best practices for the utilization of expanded carrier tests are lacking, as well as regulatory oversight of expanded carrier test companies.28–30 Consideration of couple screening is largely ignored.
Scientific guidelines on carrier test establishing the selection criteria for genes to be screened for.

ACOG: American College of Obstetricians and Gynecologists; ACMG: American College of Medical Genetics and Genomics.
There is no family history in approximately 88% of carriers 31 and therefore guidelines are evolving to highlight the importance of offering screening to all couples planning a pregnancy. Healthcare practitioners’ perceptions of factors influencing the implementation of carrier testing have been published. 32
In the vast majority of countries, screening for the general population is only available on a user pays basis, apart from haemoglobinopathy screening, which is commonly performed via a full blood examination. Individuals should be informed that there is an out-of-pocket expense: the current average individual cost of $400 US dollars implies a substantial financial barrier for many couples. The responsible and ethical implementation of genomic advances in medicine requires equal access for all couples regardless of socioeconomic factors.
Couple carrier testing for autosomal recessive disorders is highly effective and should be adopted as part of pre-pregnancy counselling and prenatal care. It has significant clinical advantages over carrier, or stepwise, screening. We recommend that advisory bodies give consideration to the advantages of couple screening when updating the clinical guidelines they produce.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article