
Abstract
Objective
To evaluate the feasibility and effectiveness of a preventive programme for haemoglobinopathies in a single centre in Northeastern Iraq.
Methods
Premarital screening, genetic counselling and prenatal diagnosis (PND) were implemented over a 5 year period.
Results
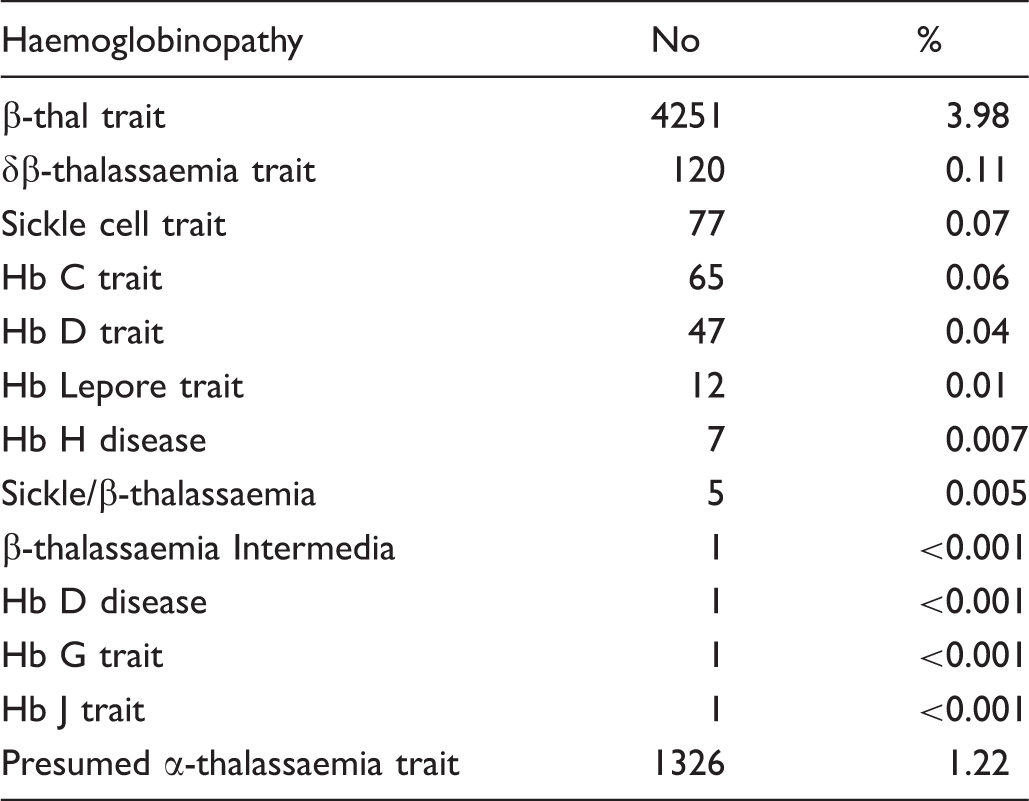
Among a total of 108,264 screened individuals (54,132 couples), β-thalassaemia trait, δβ-thalassaemia trait, and sickle cell trait were diagnosed in 3.98%, 0.11% and 0.07%, respectively. Of 130 at risk couples (2.4/1000), 107 (82%) were available for follow up, with 105 couples (98.1%) proceeding with their marriage after counselling. In the 125 registered pregnancies in the latter couples, PND was performed in 85 (in 80 couples, uptake 76%). Selective termination was chosen in 10 of the 11 pregnancies with an affected fetus. Six affected babies were born among couples who declined PND. At the same time 30 already married couples with at least one thalassaemic child underwent PND, revealing three affected fetuses; all three pregnancies were terminated.
Conclusion
The programme revealed that most at risk couples diagnosed by premarital screening chose to proceed with their marriage, with 76% seeking PND followed by selective termination of an affected fetus. A 65% reduction in number of affected births was reported over the 5 year period. This regional programme could serve as a prototype for a national haemoglobinopathy prevention programme.
Introduction
Haemoglobinopathies are autosomal recessive inherited disorders of globin chain synthesis, that may either be quantitative (thalassaemias) or qualitative (Sickle cell, Hb C, Hb E, and others). 1 They are the most frequent single gene disorders worldwide, and particularly in the Eastern Mediterranean region, including Iraq. 2 The first reports on thalassaemia major and sickle cell disease from Iraq appeared in the 1960s.3,4 Thereafter, these inherited disorders became increasingly recognized as important health problems. The carrier rates for β-thalassaemia ranged from 3.7–4.5% in various parts of the country, with some 15,000 registered patients with β-thalassaemia major/intermedia.2,5–7 Sickle cell disorders were seen mainly in the extreme north and south of the country, with the highest carrier rates in the south reaching up to16% in some localities.8,9 Such high carrier rates coupled with around a 30% rate of consanguineous marriages, 10 and the limited resources available to the health services, make initiating a preventive programme for these disorders a necessity. 11
A prevention programme based to a great extent on the Cypriot model 12 (premarital screening, genetic counselling and prenatal diagnosis [PND]) was initiated in northern Iraq in 2008, following pilot studies to determine the service indicators for prevention and defining the molecular basis of β-thalassaemia in the region.7,13–15 Furthermore, it was made legally mandatory to perform premarital screening for haemoglobinopathies, and various religious scholars agreed on the principle of allowing selective termination of the affected fetus prior to 16 weeks of gestation. 11
We here report the progress of the haemoglobinopathies prevention programme over the past 5 years in a single regional centre in Northeastern Iraq.
Subjects and methods
Subjects
All couples attending the premarital provincial haemoglobinopathy screening centre in Sulaimaniyah, Northeastern Iraq (Figure 1), which is the only centre authorized to perform such testing in the province, from 1 January 2008 to 30 December 2012 were included in this report.
Map of Iraq showing Sulaimaniyah province (shaded).
Design of the preventive programme
Premarital screening
A 5 ml blood sample was taken from each partner in the couple attending the screening centre and added to ethylenediamine tetra-acetic acid (EDTA) tubes (2 ml) and plain tubes (3 ml). The EDTA sample was first used to perform the sickling test, then processed in a haematology analyzer (Beckman Coulter, USA) (calibrated daily) to determine the red cell indices. If the mean corpuscular volume (MCV) was less than 80 fL and/or the mean corpuscular haemoglobin (MCH) less than 27 pg and/or the sickling test was positive for either member of the couple, the sample was processed further for the quantitation of Hb A2, Hb F, Hb S (if sickling is positive) or any other variants, in an automated ion-exchange high performance liquid chromatography (HPLC) system on the Bio–Rad variant instrument (Bio-Rad Laboratories, Belgium).
Subjects were labeled as β-thalassaemia minor if they had hypochromia and/or microcytosis with an HbA2 level more than 3.5%. An increased Hb F level of 5–20%, with no excess in Hb A2 level, was labeled as δβ-thalassaemia trait. Other haemoglobinopathies were documented based on HPLC results. 1 Serum from subjects with reduced MCV and/or MCH and no excess of Hb A2 or F was used to determine serum Iron and total Iron binding capacity (Biolab-France) and to calculate transferrin saturation. Among this subgroup of subjects, those with a transferrin saturation of less than 15% were labeled as positive for Iron deficiency, while those with a transferrin saturation ≥15% were given a presumptive diagnosis of α-thalassaemia.
A couple was considered to be at risk of having affected children if both partners were carriers of β-thalassaemia, or a structural Hb disorder, or if one was a β-thalassaemia carrier and the other a structural haemoglobinopathy carrier.
Counselling
Couples at risk were counselled together by a public health specialist trained in haemoglobin disorders at the premarital centre. Couples were briefed on the risk involved in proceeding with marriage, and provided with detailed information on the haemoglobinopathy from which their children may suffer. Their available options, including separation, having a limited number of children, or prenatal diagnosis during pregnancy were explained, allowing them to make an informed decision on which option to pursue. Couples signed a declaration stating that they had been counselled and understood the risk implications for their future offspring. A report was then issued by the centre to the court, stating that premarital testing for haemoglobinopathies was performed, without specifying whether the couple was at risk, to protect the privacy of the couple and their informed decision.
Prenatal diagnosis
Couples at risk were offered the choice of having a chorionic villus sampling (CVS) at 10-13 weeks of gestation to determine whether the fetus was affected. CVS sampling was performed in collaboration with the Kariminejad-Najmabadi Pathology and Genetic centre in Tehran. Couples with an affected fetus were given the choice of termination, provided that it is performed prior to 16 weeks gestation. The PND procedure was supported financially by the local government.
Educational programme
The screening programme was accompanied by an educational programme using mass media and posters and leaflets at the premarital centre and the major health institutes in the province.
As a supplementary part of the programme, prenatal diagnosis was also offered to couples who were married prior to 2008, and had at least one child with a major haemoglobinopathy registered at the thalassaemia centre in the province.
Results
The spectrum of haemoglobinopathies
The distribution of haemoglobin disorders among 108,264 subjects screened by the premarital screening programme in Sulaimaniyah province over a 5 year period.
Couples at risk
Results of premarital screening for haemoglobinopathies.

Prenatal diagnosis
The distribution of mutations among the 80 couples at risk (160 individuals), who underwent prenatal diagnosis.

The number of couples with autozygous mutations.

PND was also offered to the parents of thalassaemia major children registered at the local thalassaemia centre, with 30 couples taking up this option. Three of the 30 pregnancies were affected and all three were terminated.
Among the recorded 155 pregnancies among “at risk couples”, there were 20 affected fetuses (12.9%), with 13 selective terminations and seven affected births. Only one couple who underwent PND and were found to have an affected fetus chose to continue with the pregnancy, because of the delay in PND. The prevention programme therefore reduced the number of affected births from 20 to 7 over a 5 year period, a reduction of 65% of affected births.
Discussion
Premarital screening programmes for thalassaemia and sickle cell disorders aim to identify asymptomatic carriers of these autosomal recessive disorders, so that they are informed and can understand their reproductive risks and available options. 16 Such screening programmes constitute the backbone of the preventive strategy for these haemoglobinopathies, a strategy which has been advocated by WHO, as major haemoglobinopathies cannot be cured, unless bone marrow transplantation is successfully performed. 17 While WHO has recommended that all such screening tests should be voluntary, 18 several countries in the Mediterranean region including Iran, Cyprus, Saudi Arabia, Bahrain, Jordan and Palestinian territories have passed laws making premarital testing for haemoglobinopathies compulsory prior to marriage.2,19 The rationale for advocating such mandatory testing by the authorities in these countries (and in our region) is the belief that the method is the most effective in decreasing the birth rate of these disorders.
In our study, β-thalassemia trait was the most commonly seen haemoglobinopathy, with a carrier rate of 3.98%. This is consistent with the figures reported by an earlier pilot study in the province and within the rates reported throughout Iraq, which range from 3.7% in extreme north to 4.6% in the extreme South.6,7,15 The prevalence rates for β-thalassaemia vary in surrounding countries, with a range of 2.0% to 5.9% in the Arab countries in the Eastern Mediterranean region to the west and south of Iraq. 2 The overall frequency is 2% in Turkey (to the north of Iraq), and 4%–10% in various regions of Iran (to the East of Iraq).20,21
Other than β-thalassaemia, haemoglobin disorders, including sickle cell trait and δβ-thalassaemia trait were not common in our study. This is consistent with the fact that sickle cell disease is uncommon among registered patients at the provincial thalassaemia centre, and that none of the couples were at risk of sickle cell anemia throughout the 5 years of the study. This may support a policy of abandoning the sickling test as an initial screening premarital procedure, and considering it only when the partners are consanguineous, have a suggestive family history, or if one of the partners is a β-thalassaemia carrier.
With a ∼4% β-thalassaemia carrier rate, it would be expected that 1/625 couples planning marriage would be at risk of having affected children because both partners are carriers (1.6/1000), however, the consanguinity rate among couples in our study was 21%, suggesting that a higher at risk rate maybe expected. In a population with 30% first cousin marriages, it is expected that the birth rate of homozygotes for a condition with 3.5% carrier frequency is 2.1 times the rate expected in a population with no consanguinity. 22 The rate of 2.4/1000 couples at risk of conceiving affected children in Sulaimaniyah is nearly half that reported in the Iraqi Duhok province and in neighboring Iran (5 and 4.5/1000), but is higher than that reported in Turkey (1.5/1000 respectively).7,23,24 The differences noted above are relevant to the prevalence rates of β-thalassaemia and sickle-cell genes in these particular populations.
The β-globin gene mutations identified in the couples at risk are consistent with those identified earlier in the province, 15 however, we identified three additional mutations: the Iranian initiation codon (ATG>ATT), the mild Mediterranean -101 (C>T), and the Lebanese codon 29 (C>T), as well as the two structural variants Hb D and J-Iran. These represent the first documented reports of these mutations from Iraq.
As would be expected, consanguinity among the couples at risk was approximately double the overall consanguinity in the series. It is important therefore to carefully check consanguineous couples for haemoglobinopathies. Three of the rare mutations were found in both partners in three couples respectively (initiation codon (ATG>ATT), IVS-II-745 and codon 29). This is also expected as consanguinity increases the expression of homozygotes for rare, rather than common, mutations. 25
Despite genetic counselling, the number of couples who decided to continue with their marriage arrangements was nearly 98%. This is quite similar to that reported in India and Saudi Arabia, where rates of 99-89.6% have been reported.26–28 The main reason reported in the Saudi Arabian study, similar to our own study, was the timing of the premarital screening. The mandatory legal procedures must be performed prior to the marriage, and most couples will undertake screening only a few days before the ceremony,and after all arrangements have been finalized, so there is significant social pressure on the families and couple. The relative lack of well established population thalassaemia education programmes in the region may also be important. Most couples do not have prior knowledge about the screening programme.
As most couples choose to continue with the marriage, the option of separation will not have an impact in reducing the number of affected babies. Because of this, PND, and possibly preimplantation diagnosis, are probably options on which to focus. In some populations (eg. Palestinian Arabs), counselling directly encouraged separation and this aspect of the programme was significant in the reduction in affected babies. In our study however, informed choice was left completely to the couple at risk. 29
One option to reduce the rate of affected births may be to implement haemoglobinopathy screening among secondary school students, in parallel with an education programme, so that a decision can be made before any possible commitment to marriage. This option has been successful in some countries.30,31
Among the couples proceeding with marriage and available for a 5 year follow up, there was a 76% uptake of PND. This relatively high rate, considering that the programme was only recently introduced, indicates the effectiveness of the genetic counselling procedure. In addition, the number of pregnancies/couple was higher among couples who did not accept PND (1.6/couple) than among those who did (1.06/couple). This is consistent with the notion that the latter category of couples were better informed and had a higher level of awareness of the risks associated with their pregnancies, tending to have better planned pregnancies and opting for fewer children to reduce the risks. These aspects of our results require further scrutiny to optimize the genetic counselling towards achieving the ultimate goal of reducing the number of affected births.
Among 14 couples who chose to undertake PND and had pregnancies with affected fetuses, 13 chose to terminate (93%). The only couple who continued with the pregnancy did so because of the delay in PND results to around 16 weeks, after which termination is not possible. The almost universal choice to terminate by the couples with affected fetuses is consistent with that reported from many countries,26,30,32–34 with rare exceptions. 35
Improvements to our programme are still needed, including establishing better contact with the couples at risk (with periodic follow up), improving the genetic counselling to ensure a higher uptake of PND, upgrading and intensifying the population education programmes, offering voluntary haemoglobinopathy screening to couples married prior to 2008, and establishing better awareness among the thalassaemic families of the value of PND to prevent the birth of further affected children.
Conclusion
This study has demonstrated that a preventive programme for haemoglobinopathies based on the concept of premarital screening, counseling, and PND is feasible in the setting of a developing country such as Iraq, and is a viable way to reduce the birth of affected babies. Over the 5 years of the study, the prevention programme succeeded in lowering the number of births affected by haemoglobinopathies by 65%. This regional preventive scheme may serve as a prototype worth considering in the setting of a proposed national programme.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Acknowledgments
We acknowledge the technical assistance of the scientists at the premarital screening centre in Sulaimaniyah throughout the five years of the programme, particularly Mr H Abdul-Wahid, Ms F Aref and D. Jalal. We also acknowledge the cooperation of Dr L Rassul, the director of the thalassaemia centre at Sulaimaniyah.