
Abstract
Objective
To report the diagnostic challenges of newborn screening for abnormal haemoglobins.
Setting
Cord blood samples from 13 hospitals in southwest Jamaica taken in 2008–2019.
Methods
Blood spots, collected from the umbilical cord, were analysed by high pressure liquid chromatography (HPLC) to reveal phenotypes for HbSS and HbCC, but genotype confirmation may require parental studies or gene sequencing. Such cases that were successfully traced were analysed in this follow-up study.
Results
HPLC screening of 121,306 samples detected HbAS in 11,846 (9.8%), HbAC in 4508 (3.7%) and other electrophoretic abnormalities in 1090 babies. Among 101 previously unconfirmed cases, 34/90 (38%) with HPLC evidence of a HbSS phenotype had other genotypes, and 7/11 (64%) with a HbCC phenotype had other genotypes. Syndromes from the interaction of β thalassaemia occurred in 112 babies (85 with HbS, 27 with HbC) and of genes for hereditary persistence of fetal haemoglobin (HPFH) in 18 (12 with HbS, 6 with HbC). Variants other than HbS and HbC occurred in 270 babies, 16 in combination with either HbS or HbC, and 254 as traits. Most variants are benign even when inherited with HbS, although HbO Arab, HbD Punjab, or Hb Lepore Washington, which occurred in 6 cases, may cause sickle cell disease.
Conclusions
Genes for β thalassaemia and HPFH are common in western Jamaica and when associated with HbS may present diagnostic challenges in newborns, as HbF and HbA2 have not reached diagnostic levels. Family and DNA studies may be necessary for genotype confirmation.
Introduction
The Manchester Project, set in the parish of Manchester in central Jamaica, offered free, voluntary screening for haemoglobin genotype to the senior classes of 15 secondary schools. 1 To determine whether knowledge of haemoglobin genotype influenced choice of partner and the prevalence of sickle cell disease among their offspring, newborn screening was set up in hospitals in southwest Jamaica where the female students were most likely to have their offspring. Results of this newborn screening up to March 31, 2015 2 and the effect of this knowledge on early reproductive outcome 3 have been presented. By the end of 2019, a total of 121,306 babies had been screened and, after excluding 13 early deaths and 3 families declining investigation, there were 140 cases in whom preliminary findings on high pressure liquid chromatography (HPLC) suggested significant disease but this had not been confirmed. This study presents the results in 101 of these 140 cases who were successfully traced and adds information on molecular variants. The practical issues encountered in this predominantly rural population may be of value to others setting up similar programmes.
Material and Methods
Patient population. Newborn screening commenced in Mandeville Regional Hospital on August 4, 2008, extending to Percy Junor Hospital in Spalding, and a small private hospital (Hargreaves Memorial) in Mandeville in January 2009, then to May Pen and Black River Hospitals in January 2010. This screening proved efficient and cost-effective and the service was offered to the Western Healthcare region, commencing with Savanna-la-Mar Hospital on December 4, 2013, Cornwall Regional Hospital in Montego Bay on March 1, 2014, Noel Holmes Hospital (Lucea) on June 17, 2014, and Falmouth Hospital on July 1, 2014. Small rural hospitals such as Lionel Town and Chapelton routed samples through May Pen Hospital, and the private Royale Medical Centre in Savanna-la-Mar sent samples to the local public hospital for routing to the diagnostic laboratory in Mandeville, the capital of the parish of Manchester. Some diligent nurse/midwives also collected samples from home deliveries. Births at small private hospitals in Montego Bay (Montego Bay Hospital, Hospiten) also used this diagnostic service but the results are excluded because of atypical features of their populations. The current report covers the 12-year period August 4, 2008 to December 31, 2019.
On first contact, usually by phone or interview, details of relatives and their contact data were obtained in case of failure of phone service with the mother. Mothers uncontactable by phone received home visits from staff of the sickle cell programme or public health nurses, and if this failed the nearest police station was requested to assist tracing the mothers. The programme was arranged in collaboration with the Ministry of Education and the Ministry of Health & Wellness and was transferred to the latter Ministry in July 2017.
Preparation for the screening programme. All hospitals received preliminary lectures on sickle cell disease emphasizing its genetics, the importance of early diagnosis, and the techniques for sample collection. Blood spots, on specially prepared Guthrie cards, were collected as the umbilical cord was cut or by heel-prick before discharge if cord samples were missed, audits indicating collection rates of 98.5%. After the blood spot was dried, the cards, which bore identification and contact data, were batched and forwarded to the diagnostic laboratory in Mandeville. To keep the staff informed, newsletters were sent every 3 months summarizing the overall progress of the study, the performance of the individual hospitals and the results of sample analysis.
Establishment of the central laboratory and sample investigation. Based at the offices of the Southern Regional Health Authority in Mandeville, the laboratory provided facilities for haemoglobin electrophoresis on cellulose acetate and agar gel, haematology analysers, HPLC (initially Bio-Rad Variants and later the NBS model), and a microscope for the slide sickle test As cord blood samples arrived, a section of the blood spot was cut, eluted in distilled water, and analysed by HPLC. Peaks in the positions of HbS or HbC and absent or trace amounts in the HbA window were suspected to be clinically significant genotypes and families were requested to attend for confirmation and parental studies.
Parental samples were examined by alkaline haemoglobin electrophoresis and for red cell indices. HbS on electrophoresis was confirmed by the slide sickle test and HbC by agar gel electrophoresis at pH 6.2. Negative confirmatory tests for HbS and HbC were investigated by genomic sequence analysis 4 of HBB, HBA1 and HBA2 at the Weatherall Institute of Molecular Medicine, Oxford, UK or the Department of Paediatric Oncology, Hematology and Immunology, University of Heidelberg, Germany and identified using the syllabus of haemoglobin variants. A HbAA pattern associated with a mean cell haemoglobin ≤ 26 pg and red blood cell distribution width (coefficient of variation) [RDW(CV)] < 18.0 were considered candidates for β thalassaemia trait and had estimations of HbA2 performed. Samples with HbA2 ≥ 3.5% underwent DNA sequence analysis of polymerase chain reaction-amplified β-globin DNA 5 in the Department of Paediatric Oncology, University of Heidelberg, Germany. Samples with > 8% HbF, measured by HPLC, were investigated by multiplex ligation-dependent probe amplification (MLPA, MRC-Holland, Amsterdam, The Netherlands) to identify deletional hereditary persistence of fetal haemoglobin (HPFH) or by DNA sequencing of gamma globin promoter regions for non-deletional HPFH at the Red Cell Centre, Kings College Hospital, London, UK.
Allocation of haemoglobin genotype. A dominant peak in the position of HbS with minimal traces in the HbA window was consistent with HbSS, which was confirmed, if both parents showed the HbS gene, by HbA2 levels of ≤ 4.4% or by sequencing of DNA. HbS or HbC with β thalassaemia, HPFH, or other variants, suspected from family studies, were always confirmed by DNA sequence analysis. HbSC was confirmed if both HbS and HbC had positive confirmatory tests, but family studies were pursued to provide information for counselling purposes. DNA confirmation was not considered necessary. Studies to confirm genotype were performed before the baby was 6 weeks old to allow referral to a local clinic for follow-up and pneumococcal prophylaxis, if indicated.
Statistical Methods Gene frequency calculation was restricted to β chains and performed by standard gene counting methods. Gene frequencies were compared using Poisson regression. As not all the 254 variant traits were identified, gene counting was limited to confirmed variants and assumed that alpha and gamma chain variants contributed 2 HbA genes and β chain variants contributed 1 HbA gene to the gene pool.
Results
These are presented in two sections, firstly the investigation of unconfirmed cases in the dataset which closed on 31 December 2019 and then a summary of the overall genotype findings.
Unconfirmed genotypes. There were 121,306 newborns screened in 13 hospitals and, of these, 140 babies lacked genotype confirmation. A total of 39 could not be traced: 3 now lived outside the area, 3 had equivocal results on DNA and could not be traced for further investigation, 6 had never been contactable, and 27 failed repeatedly to keep appointments. Of the 101 traced cases, provisional diagnosis by HPLC suggested an HbSS phenotype in 90 cases and a HbCC genotype in 11 cases. Confirmation of the 90 HbSS cases revealed that 34 (38%) had other genotypes (25 HbS/β+ thalassaemia, 3 HbS/βo thalassaemia, 3 HbS/HPFH, 3 HbAS). Of those suspected to have HbCC, 7 (64%) had other genotypes (4 HbC/β+ thalassaemia, and single cases of HbC/HPFH, HbC/Hb Camden, and HbAA).
Final results of the completed dataset. Of the 13 hospitals contributing newborns, 8 principal hospitals with the number of sampled deliveries are shown geographically in Figure 1. Screening detected HbAS in 9.8%, HbAC in 3.7%, and other electrophoretic abnormalities in 1090 or 0.90% babies (Table 1). The basis for genotype confirmation of HbSS was the demonstration of the HbS gene in both parents in 238/325 (74%), and supporting the diagnosis of HbS/β thalassaemia, HbS/HPFH or HbS/variant in 77/110 (70%). Family studies of suspected HbSS were inconclusive in 87 (fathers unavailable in 61, had inconsistent genotypes in 22, and 4 parents had sickle cell-β+ thalassaemia or HbS/HPFH so could have given either abnormal gene to their offspring), and in these the genotype was confirmed by compatible HbA2 levels or DNA sequencing. There were 112 babies in whom β thalassaemia genes were inherited with HbS or HbC, and of these 85 (76%) were of the mild β+ type and 19 had the βo thalassaemia genes, of which the IVSII-849 A > G accounted for 10/19, with 7 other molecular mechanisms (Table 2).
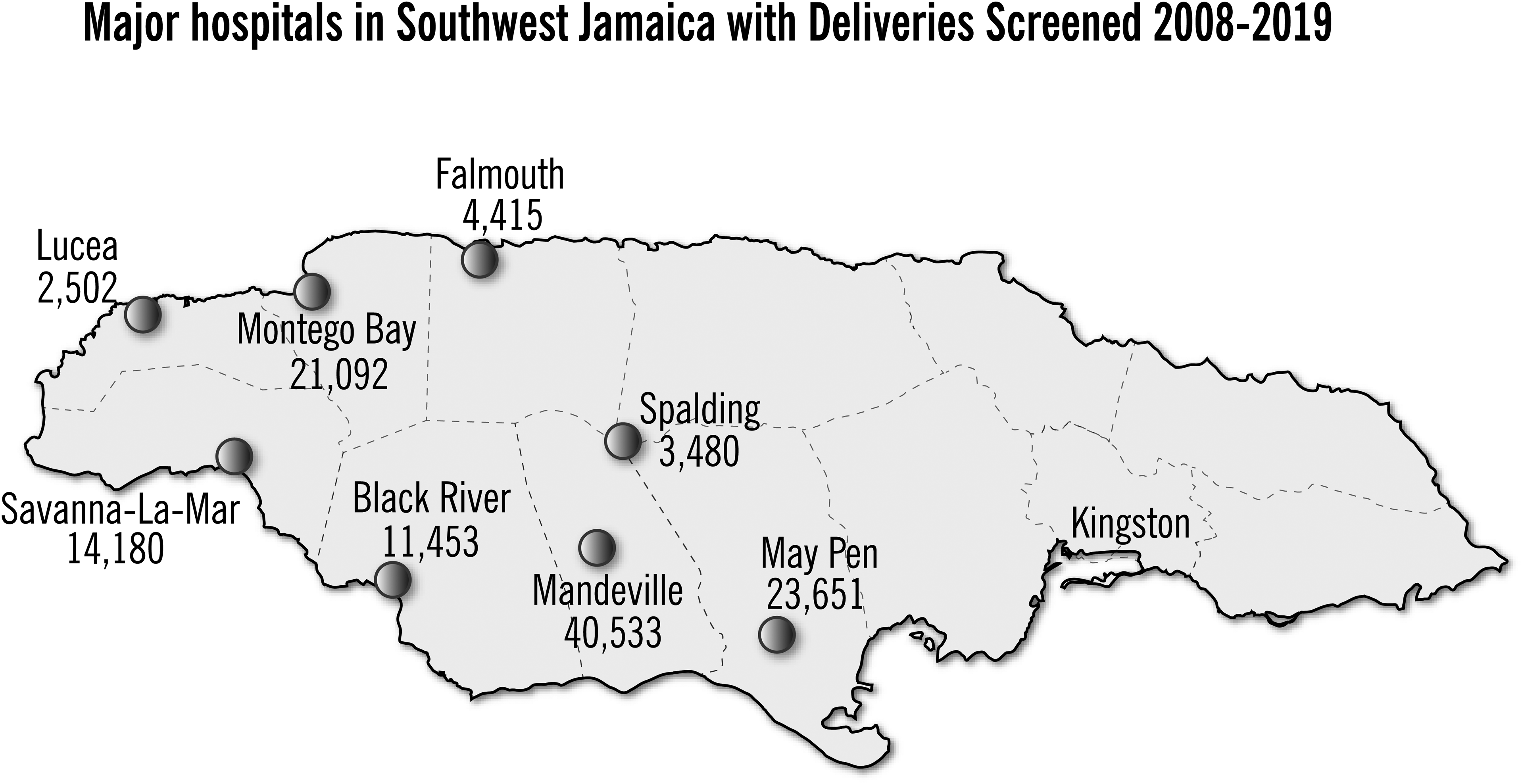
Major hospitals in southwest Jamaica with deliveries screened 2008–2019.
Results in 121,306 newborns in southwest Jamaica.
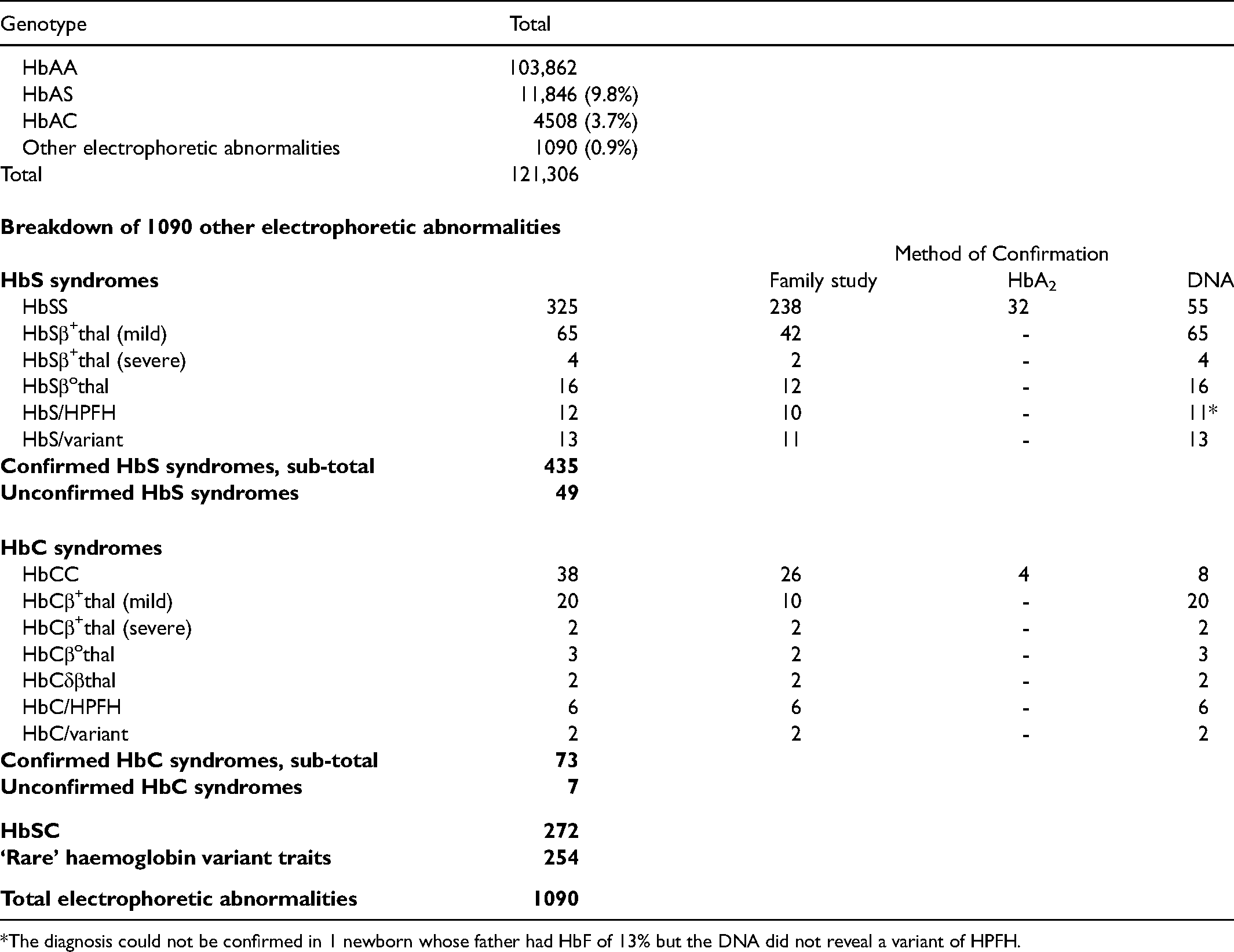
The diagnosis could not be confirmed in 1 newborn whose father had HbF of 13% but the DNA did not reveal a variant of HPFH.
Molecular variants causing beta thalassaemia.
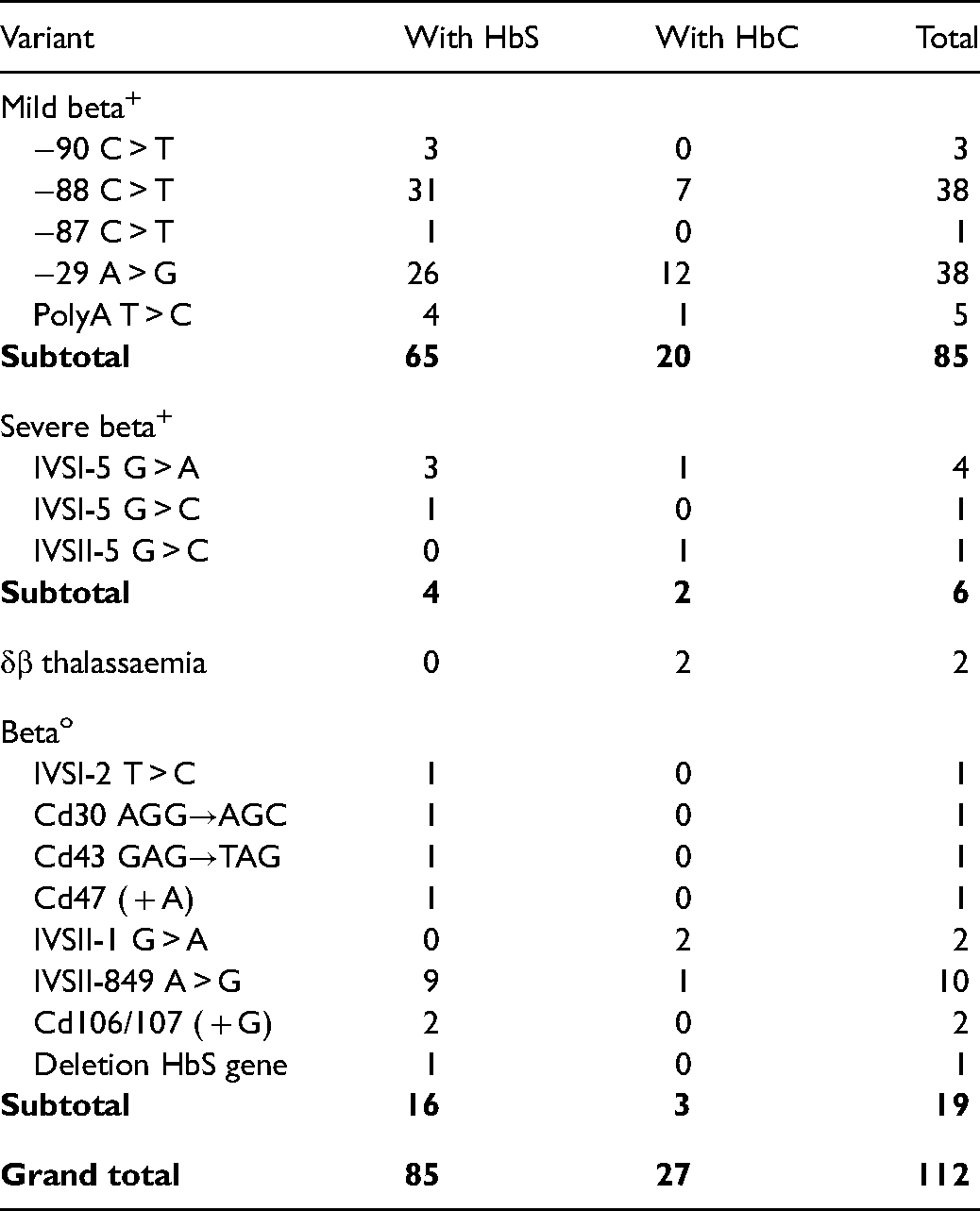
Rare variants, defined as those other than HbS and HbC, occurred as traits in 254 and as double heterozygotes in 16 (see Appendix 1). Although only 4 suspected gamma chain variants were confirmed by sequencing, gamma chain variants were considered highly likely in 26 additional families where both parents had an HbAA phenotype and the variant band disappeared with age. Most common among the β chain variants was Hb Osu Christiansborg, which does not cause disease when inherited with HbS, but inheritance of HbS with HbO Arab, HbD Punjab, or the delta/β fusion gene Hb Lepore Washington, which occurred in 6 cases, may cause sickle cell disease. The combination of HbG Philadelphia with HbS is totally benign.
Discussion
Accurate diagnosis of haemoglobin genotype is essential for appropriate management. The more severe genotypes such as HbSS and possibly HbS/βo thalassaemia lose splenic function early in life rendering patients prone to overwhelming septicaemias. Pneumococcal prophylaxis can prevent or ameliorate many infections and traditionally starts at age 2 months with conjugate vaccines and prophylactic penicillin. In developing societies, these are not only expensive but difficult to monitor and may not be justified unless there is clear evidence of benefit. The diagnosis of HbS/HPFH is relevant for two reasons, the confusion which may result for the affected individual 1 but also the lack of necessity for prophylaxis in a condition which is usually asymptomatic and with presumed normal splenic function. The role of pneumococcal prophylaxis is less clear in the mild forms of HbS/β+ thalassaemia with 15–25% HbA,6,7 which inhibits polymerization, lead to a mild clinical course, and probably allow splenic function to persist, as in Indian patients with 15–25% HbF. 8 On a literature review, the authors were unable to find records of pneumococcal septicaemia in the mild forms of sickle cell-β+ thalassaemia and so the costs and inconvenience of pneumococcal prophylaxis may be unnecessary. Far from being a rare condition in Jamaica, there is a need for proper clinical and haematological documentation partly because its benign course ensures that many cases do not present to medical attention with the result that it is perceived to be uncommon and the observed cases are biased by presenting symptoms.
The value of family study is again confirmed by these data. Often family studies are not pursued elsewhere because of the concerns related to inconsistent genotype in the father, but fathers revealed the HbS gene in 74% of babies with HbSS and only 22/260 (8%) fathers showed inconsistent genotypes, most likely resulting from impaternity, not dissimilar from the 11% impaternity recorded in the Jamaican population. 9 The other major finding is the frequency of other genotypes in cases where HPLC suggested HbSS or HbCC. The frequency of traits for HPFH in Jamaicans at 0.33% 1 is higher than the 1 per thousand often quoted in African-Americans10,11 and there has already been a plea for detection of HbS/HPFH at birth. 12 In Jamaica, the syndrome of HbS/β+ thalassaemia was detected in 35 babies screened at Victoria Jubilee Hospital in Kingston for a prevalence of 0.035% 13 and occurred in 65 (0.054%) babies in southwest Jamaica. Previous observations indicated that the clinic operating in Montego Bay in the west of the island, although accounting for approximately one-tenth of the national database known to the Sickle Cell Unit in Kingston, accounted for one-third of the cases of sickle cell-β thalassaemia, 14 suggesting that the β thalassaemia gene is more common in the west of the island. The current study confirms a higher frequency of both β+ thalassaemia and HPFH in this area.
Haemoglobin variants other than HbS and HbC were common. Hb Osu Christiansborg, which accounted for 44% of β chain variants, appeared to have a locus around the Montego Bay area, and does not cause haematological abnormalities when inherited with HbS. The variants HbO Arab, HbD Punjab, and HbLepore Washington interact with HbS, sometimes causing serious symptoms, so their identification is important in newborn screening programmes. The condition HbSE appears generally benign although symptoms have been reported in some cases. 14
In summary, the diagnosis of sickle cell-β+ thalassaemia and of HbS/HPFH in the newborn period may be complicated, as HbF and HbA2 have not reached diagnostic levels. While recognizing that both these genes may be more common in Jamaicans, it is hoped that these observations may be useful in newborn screening among Jamaicans and by inference the large Jamaican diaspora, and in West Africa from where the Jamaican population is derived.
Footnotes
Acknowledgements
We thank the nurses and midwives of the hospitals in southwest Jamaica for their interest and diligence in sample collection and the doctors in the region who collaborated in tracing patients. We thank the staff of the Southern Regional Health Authority, Ministry of Health & Wellness and Dr Michael Coombs, the then Technical Director for hosting the diagnostic laboratory in Mandeville. SLT received funding from the Medical Research Council while at Kings College Hospital, London. We thank Mrs. Christine Moore-Fuller of IRP Production, Jamaica for the Figure.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Health Fund of Jamaica, Alcoa Foundation, (grant number HPP70).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.