
Abstract
Objectives
To evaluate the feasibility of a newborn screening and follow-up programme for sickle cell disease (SCD) among tribal populations of south Gujarat, India.
Methods
A total of 5467 newborn babies were screened over 2 years using High-performance liquid chromatography, with diagnosis by molecular analysis. The SCD babies were followed-up clinically and haematologically regularly for 1.5 to 5 years to describe the course of the disease.
Results
Thirty-three babies (0.60%) were sickle homozygous, 13 (0.23%) were-sickle-β-thalassaemia, 687 (12.5%) were sickle heterozygous, and 4736 were unaffected. The parents of SCD babies were educated and counselled for home care. There were 32 babies (69.5%) who could be clinically and haematologically followed-up; 7 babies (21.8%) presented with severe clinical complications, whereas 18 (56.2%) babies were asymptomatic till the last follow-up. The variation in clinical presentation was seen in spite of the presence of ameliorating factors, such as high fetal haemoglobin, Xmn-I polymorphism, and α-thalassaemia.
Conclusion
In addition to demonstrating the possibility of establishing a newborn screening programme for sickle cell disorders among tribal populations, this study has shown that the disease is not always mild among tribal groups in India, as previously believed. There is a need, therefore, for increasing awareness among these tribal groups about the disease, and for regular monitoring of affected babies to reduce morbidity and mortality and to understand the natural course of the disease.
Introduction
Sickle cell disease (SCD) is the most common haemoglobinopathy in the world. 1 Affected infants generally present clinically with painful swelling of the hands and feet (dactylitis), pneumococcal sepsis or meningitis, severe anaemia and acute spleen enlargement, acute chest syndrome, pallor, jaundice, or splenomegaly. Damage affecting the kidneys, bones, and joints, and chronic pain and disability, which compromise the quality of life, are the long-term consequences of SCD, together with the possibility of early sudden death related to the disease. 1 Newborn screening for haemoglobinopathies, now routine practice in many parts of the world, 2 identifies infants with SCD who can be provided with early comprehensive care to reduce morbidity and mortality. In the United States it is currently available in all 50 states and the District of Columbia.3–4 Pilot programmes have been established in the Philippines, Brazil, Ireland, Saudi Arabia, Spain, Burkina Faso, Senegal, United Arab Emirates, Rwanda, England, Democratic Republic of the Congo, Benin, Netherlands, Angola and recently a newborn screening programme has also been started in Mali.5–27 In spite of this there have been only two preliminary reports on newborn screening from India, where SCD is prevalent, and a major health burden.28,29
In India, sickle cell anaemia is mainly seen among tribal populations in western, central, eastern and some parts of southern India and in a few non-tribal communities where the prevalence of carriers ranges from 2% to 34%.30,31 Precise figures on annual SCD births in India are unavailable, but newborn screening for SCD and other haemoglobinopathies has a significant potential public health impact. The aim of this study was to evaluate the feasibility of establishing a newborn screening and follow-up programme for sickle cell disorders in the rural tribal regions of south Gujarat.
Methods
A newborn screening programme among tribal populations was implemented in four districts in south Gujarat (Valsad, Navsari, Dang, and Surat), with samples collected from 13 different centres (Figure 1).
Map showing major collection centers in south Gujarat.
Education and awareness programmes were conducted for 143 medical officers from primary health centres, community health centres, and district hospitals, for private medical practitioners, including 30 gynaecologists and paediatricians, as well as multipurpose health care workers in these areas. Training in newborn heel prick sampling, and clinical evaluation of the babies during follow-up was provided. Booklets, pamphlets and videos in the local language were also distributed to centres.
Informed parental consent was obtained before sample collection. Babies were screened either at birth or soon after (up to 1 month) in the majority of cases. Blood samples were collected by heel prick on Whatman filter paper S & S 903, and in a subset of babies also from the umbilical cord in EDTA vacutainers.
The EDTA vacutainers or the dried blood spots were transported to the Valsad Raktadan Kendra, Valsad within 24 hours of collection. Filter paper samples were analyzed by High-performance liquid chromatography (HPLC) using the Newborn Screening machine (Bio-Rad Laboratories, Hercules, CA) according to manufacturer instructions. The EDTA samples were run on the Variant Hemoglobin Testing System (Bio-Rad Laboratories, Hercules, CA), to cross check the results from the Newborn Screening machine as this was the first time the machine was being used in India.
Molecular analysis of all presumptive sickle homozygous/sickle-β-thalassaemia cases, and few sickle heterozygous and normal babies randomly selected, was performed during the follow-up for confirmation of diagnosis at National Institute of Immunohaematology, Mumbai, by amplification refractory mutation system (ARMS). 32 The −158 (C→T) nucleotide change (Xmn I polymorphism) in the Gγ globin gene promoter was studied by PCR and restriction enzyme digestion 32 ; α-genotyping for eight deletions was performed using multiplex PCR. 33
The sickle homozygous and sickle-β-thalassaemia babies were registered at the sickle cell clinic at Valsad for follow-up at an interval of 3–4 months. Pneumococcal vaccination and folic acid supplementation were given to all babies. During follow-up, the babies were clinically evaluated and complete blood count and HPLC analysis was performed. Parents and siblings were also investigated. Parents were counselled about the inheritance and possible clinical complications in their SCD child. Educational material on SCD, newborn screening, and the possibility of prenatal diagnosis in subsequent pregnancies was presented in brochures, videos and DVDs. These were also presented at multiple community events and health fairs. The parents of these SCD babies were given information on home care, and were educated to detect symptoms that may lead to serious medical emergencies. The study was approved by the Institutional Ethical Committees of Valsad Raktadan Kendra, National Institute of Immunohaematology and by the Institutional Review Board of the University of Pittsburgh Medical Center.
Results
Prevalence of sickle cell trait and sickle cell disease babies in different tribal groups.
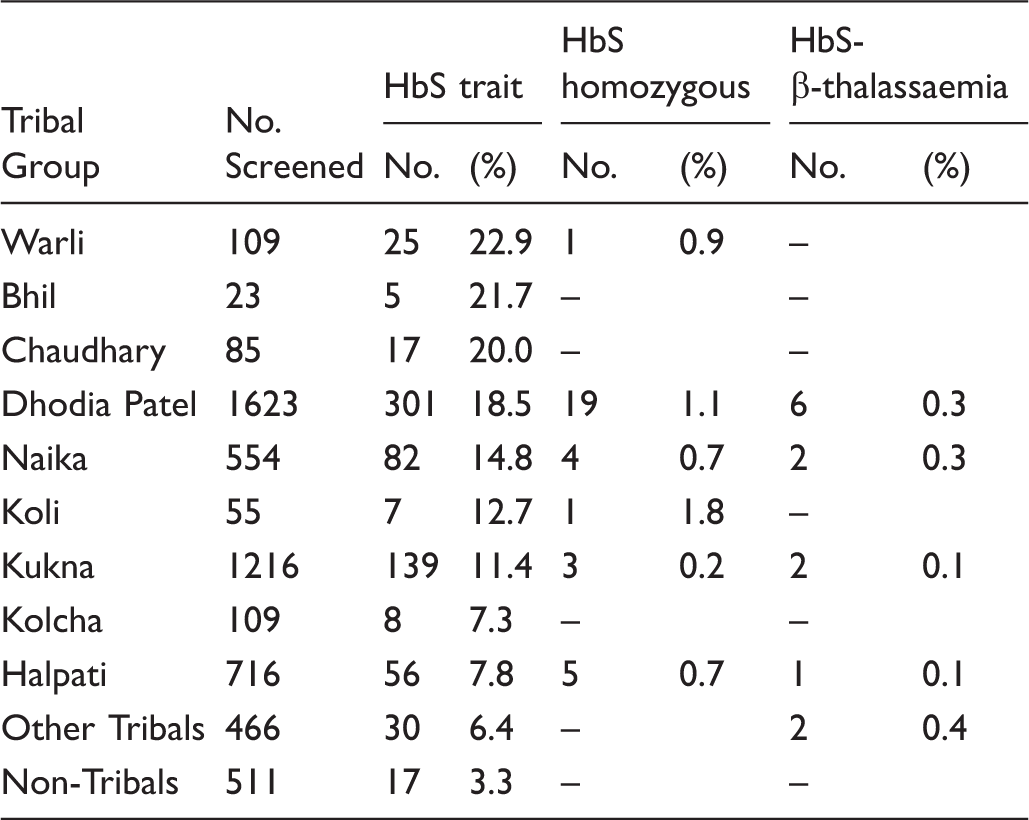
Despite multiple attempts to contact the parents of babies identified with SCD, only 32 of the 46 affected babies (69.5%) could be followed-up regularly for clinical and haematological evaluation. Pneumococcal vaccination (23-valent polysaccharide vaccine-PNEUMOVAX® 23) was given to the children who were regularly followed-up. Seven families could not be contacted due to incorrect addresses and telephone numbers. Three families refused confirmatory testing and follow-up, as they believed their children were healthy. Four couples followed-up at the sickle cell clinic with their child only two or three times till their children were aged 3 months to 1 year.
Table 2 summarizes the clinical presentation of the 32 babies who were regularly followed-up to age 1.5–5 years. Of these 32 babies, 18 (56.2%) (11 sickle homozygous and 7 sickle-β-thalassaemia babies) had no clinical complications till the last follow-up. The age at first clinical presentation of the others varied from 1 month to 5 years. Seven babies (21.8%) (6 sickle homozygous and 1 sickle-β-thalassaemia babies) presented with severe clinical complications, such as sepsis and severe vaso-occlusive crisis requiring frequent hospitalization, and anaemia, requiring blood transfusion; six other patients presented with severe anaemia and fever with infection, but their symptoms were moderate and did not require hospitalization (Figure 2). One sickle homozygous baby had Down’s syndrome, and another presented with mental retardation and hypotonia. Complete blood counts conducted during the last follow-up yielded haemoglobin levels ranging from 7.0 to 10.5 g/dl among the sickle homozygous babies, and from 7.5 to 10.4 g/dl among the sickle-β-thalassaemia babies. Mean corpuscular volume ranged from 60.2 to 88.7 fl among sickle homozygous babies, and 58.8 to 68.0 fl among sickle-β-thalassaemia babies, and mean corpuscular haemoglobin levels ranged from 18.0 to 28.2 pg among sickle homozygous babies and 16.9 to 21.3 pg among sickle-β-thalassaemia babies. The mean corpuscular haemoglobin concentration varied from 29.9 to 33.9 g/dl among the sickle homozygous babies, and from 29.6 to 33.5 g/dl among sickle-β-thalassaemia babies. Fetal haemoglobin levels varied from 12.5 to 30.2% among sickle homozygous babies (aged between 2.4 and 5 years) and 5.5 to 37.9% (between 2.5 and 5 years). HbA2 levels were increased in all sickle-β-thalassaemia children (>5.1%), although using HPLC, HbA2 is not always accurately measured and is sometimes falsely elevated in the presence of HbS, due to HbS adducts.
The clinical presentation of severe sickle cell disease babies during the entire follow-up period. The clinical presentation of sickle cell disease babies during the follow-up period.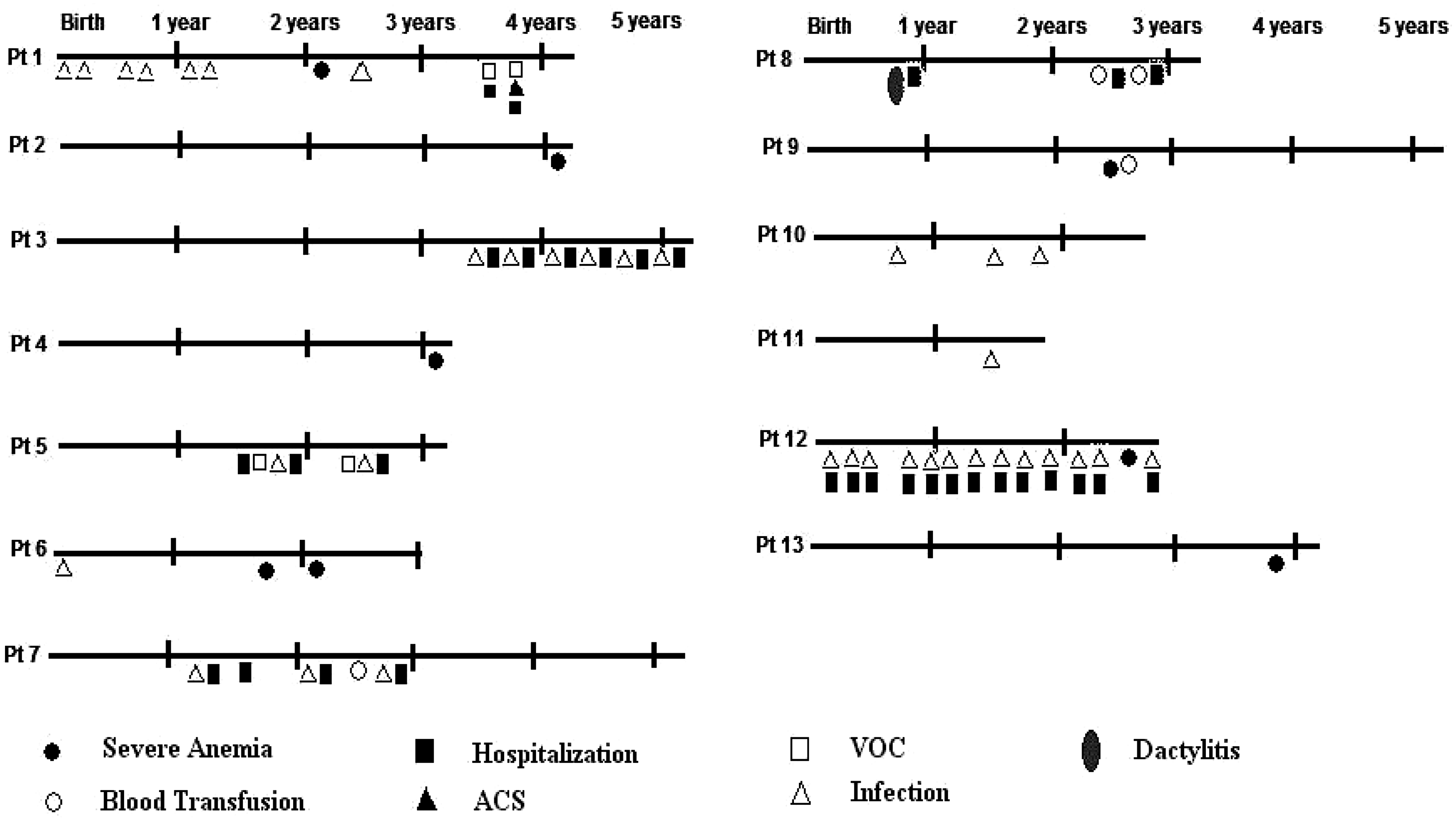
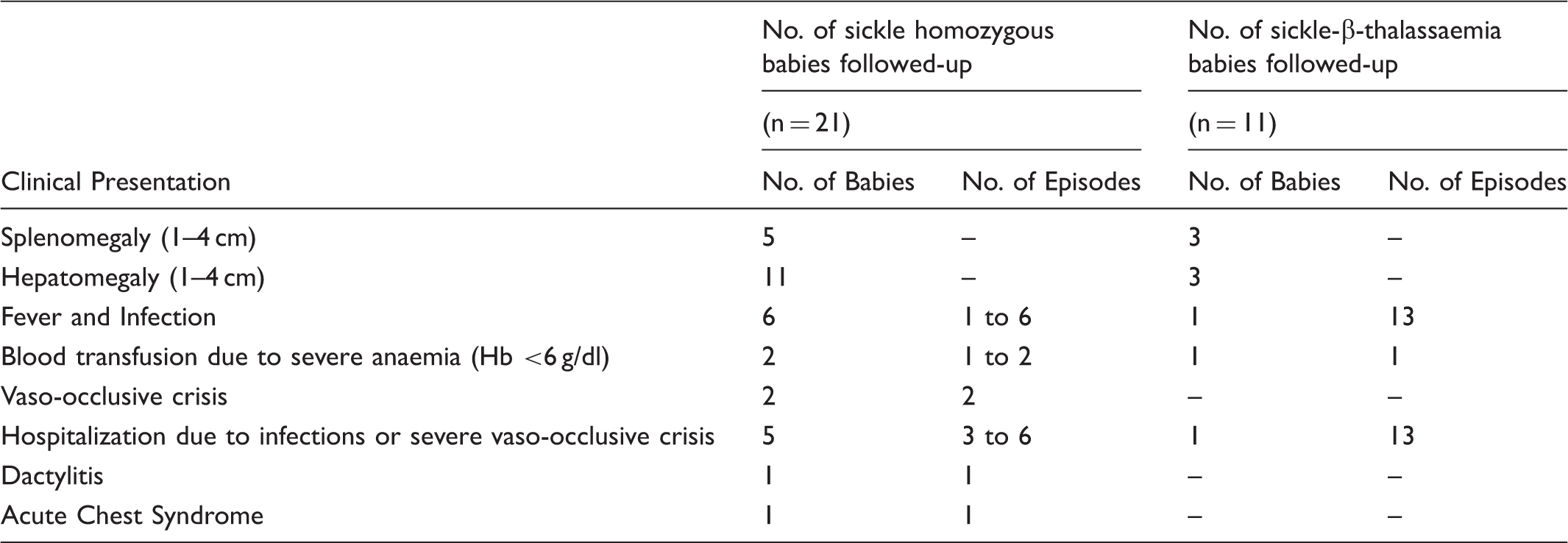
Molecular analysis for the confirmation of sickle and β-thalassaemia gene showed the presence of β-globin gene mutations among the 13 sickle-β-thalassaemia babies [β0 thalassaemia mutation Codon 15 (G→A) {HBB:c.48G > A} in 11 and severe β+ thalassaemia mutation IVS 1–5 (G→C) {HBB:c.92 + 5G > C} in 2]. All 24 sickle homozygous babies screened for Xmn I polymorphism were Xmn I (+/+), whereas of the 13 sickle-β-thalassaemia babies screened, all were Xmn I (+/−). Among the 24 sickle homozygous babies screened for α gene deletions, 22 (91.7%) had α-thalassaemia (−α3.7/−α3.7, n = 19; −α3.7/−α4.2, n = 1; −α3.7/αα, n = 1; −α4.2/αα, n = 1), whereas 2 babies showed no α gene deletions. Nine sickle homozygous babies who were lost to follow-up could not be screened for Xmn I polymorphism and α-thalassaemia. Twelve of the 13 sickle-β-thalassaemia babies (92.3%) screened for α gene deletions also had α-thalassaemia (−α3.7/−α3.7, n = 8; −α3.7/−α4.2, n = 1; −α3.7/αα, n = 3), whereas only one baby had no α gene deletion. The low mean corpuscular volume and mean corpuscular haemoglobin levels may be attributed to the very high prevalence of α− thalassaemia in the tribal populations of south Gujarat. The five sickle homozygous babies and two sickle-β-thalassaemia babies who presented with severe clinical complications were Xmn I (+/+) and (+/−) respectively, and all had associated α-thalassaemia (−α3.7/−α3.7 − 6; −α3.7/−α4.2 – 1).
Discussion
The objective of newborn screening for SCD is to provide early care to reduce the morbidity and mortality associated with the disease. Forty years of newborn screening in the United States suggests that successful implementation of newborn screening requires the establishment of systems for collection and transportation of dried blood specimens, analytic methods for high throughput screening and quality control, and post-analytic systems for notification of families of affected newborns. and implementation of confirmatory testing and comprehensive care relatively soon after birth. 34 We demonstrate, in this study, that it is feasible to adapt and successfully apply these methods in a developing country in a largely rural, often mobile, disadvantaged population.
Two reports from India have described newborn screening efforts in single institutions.28,29 In one study, 1158 babies were screened for sickle cell anaemia in a hospital in Chhattisgarh (using the Bio-Rad Hemoglobin Variant Neonatal sickle cell short programme); 0.4% of babies had SCD, 5.26% had sickle cell trait, and 0.08% were double heterozygous for sickle-β-thalassaemia. 28 In a second study, in Nagpur, central India, 8243 mothers who delivered live babies were screened (solubility test). The newborns of the 1178 sickle positive mothers were then screened by HPLC; 536 babies were sickle heterozygous, and 88 sickle homozygous. 29 The majority of the screened newborns in Nagpur belonged to non-tribal ethnic groups. It has been reported that SCD patients from this region have a severe presentation, and hydroxyurea was beneficial in reducing the clinical severity of the condition in these patients. 35 However, in our study, in spite of the genetic modifiers such as Xmn I polymorphism associated with high fetal haemoglobin levels and α-thalassaemia, 21.8% of SCD babies required hospitalization. This is in contrast to earlier reports in which mainly adult Indian tribal patients with SCD were shown to be clinically mildly affected, due to the presence of the Xmn I (+/+) polymorphism and a high prevalence of α−thalassaemia. This has also been observed in eastern Saudi Arabia.36–40
Ours was the first study in India to establish a regional newborn screening programme, with a network of 13 delivery centres serving as newborn screening facilities. Most deliveries in Gujarat are hospital-based, but in a few cases the samples were collected later at home visits. The study has demonstrated the feasibility of the use of dried blood spots at point of delivery in remote areas. The successful transportation of dried blood spots to the newborn screening centre for analysis is particularly notable in the largely rural area covered by this project, and is relevant to conditions in India where more than 70% of the population live in rural areas with limited access to infrastructure.
We also developed analytic systems, including primary laboratory testing with HPLC and secondary confirmatory testing using molecular methods, and established quality control protocols. We took a targeted rather than universal screening approach, because it was considered to be more cost effective to screen newborns belonging to ethnic groups with a high prevalence of the HbS gene. With expansion of newborn screening to conditions other than haemoglobinopathies, newborn screening programmes may have to consider universal screening. A major limitation of this newborn screening programme was that it did not include the establishment of comprehensive care centres for the conditions detected, other than providing Pneumococcal vaccination, regular clinical and haematological work-up, and folic acid supplementation to these babies. Penicillin prophylaxis was not started as earlier studies in adults in this region had suggested that sickle cell anaemia patients belonging to these tribal populations would only have mild infection related crises. However, this study will help to inform policy makers on the future use of penicillin prophylaxis among the newborns with SCD among these tribal groups. While follow-up was offered to patients at the sickle cell clinic, only 32 babies (69.5%) regularly attended follow-up. It is likely that the non-attenders (30.5%) were asymptomatic and that therefore parents were reluctant to follow-up, and a small number of babies may have died due to an acute adverse event. To overcome problems with follow-up, we plan to give cellular mobile phones to the parents of such children.
There is limited data on the comprehensive care delivered, or on complications or long term outcomes of newborns diagnosed with haemoglobinopathies in India. Future projects must include the establishment of centres which provide comprehensive care and capture data on long term outcome of newborns diagnosed with screenable conditions.
Our model for integrating newborn screening into a hybrid system of a government-run network and private agencies has demonstrated the feasibility of a regional newborn screening system for haemoglobinopathies and follow-up of the affected babies in a disadvantaged population in a developing country.
Footnotes
Acknowledgements
We thank the technical staff appointed on this project for their support.
Conflicting interests
The authors have no competing interest.
Funding
This work was supported by the Grant Number 63/6/Indo/-US/2007-RHN dated 1/10/2008 from Indian Council of Medical Research, New Delhi and Grant number 1R03HD057740-01 Eunice Shriver Kennedy National Institute of Child Health and Human Development.