
Abstract
Objective
To evaluate the test performance of a novel sequencing technology using molecular inversion probes applied to cell-free DNA screening for fetal aneuploidy.
Methods
Two cohorts were included in the evaluation; a risk-based cohort of women receiving diagnostic testing in the first and second trimesters was combined with stored samples from pregnancies with fetuses known to be aneuploid or euploid. All samples were blinded to testing personnel before being analyzed, and validation occurred after the study closed and results were merged.
Results
Using the new sequencing technology, 1414 samples were analyzed. The findings showed sensitivities and specificities for the common trisomies and the sex chromosome aneuploidies at >99% (Trisomy 21 sensitivity 99.2 CI 95.6–99.2; specificity 99.9 CI 99.6–99.9). Positive predictive values among the trisomies varied from 85.2% (Trisomy 18) to 99.0% (Trisomy 21), reflecting their prevalence rates in the study. Comparisons with a meta-analysis of recent cell-free DNA screening publications demonstrated equivalent test performance.
Conclusion
This new technology demonstrates equivalent test performance compared with alternative sequencing approaches, and demonstrates that each chromosome can be successfully interrogated using a single probe.
Introduction
Cell-free DNA (cfDNA) testing has dramatically altered the genetic screening landscape since its commercial introduction in late 2011. 1 Genomic microarrays and sequencing utilizing shotgun, targeted, or single nucleotide polymorphism-based technologies have allowed access to the fetal genome by analysis of cfDNA fragments from maternal blood. Following the discovery of the occurrence of fetal cfDNA in maternal plasma in 1997, 2 and the publication of two articles in 2008 demonstrating the potential utility of this observation for prenatal screening,3,4 the common trisomies (21, 18, and 13) and the sex chromosome aneuploidies became the early focus. The commercial availability of cfDNA testing then set the stage for broad clinical use in the US and abroad.5–9 Additionally, some sub-chromosomal deletions can now be characterized utilizing non-invasive cfDNA testing.10–14
Sensitivities and specificities of these cfDNA tests are high (99% for the common trisomies). Knowledge of the prevalence rate of these aneuploidies in the population being screened allows the calculation of positive and negative predictive values of the testing results. It is these metrics that are most useful to clinicians counseling their patients. 15 Because these fetal cfDNA fragments are shed from the cytotrophoblast, cfDNA screening results cannot be diagnostic, due to confined placental mosaicism and true fetal mosaicism. 16
This study aimed to detect all fetal whole chromosome abnormalities on chromosomes 13, 16, 18, 21, X, and Y, through analysis of cfDNA in maternal blood, utilizing a novel sequence targeting approach using molecular inversion probes (MIPs).
Methods
The decrease in prenatal invasive diagnostic testing due to the success of cfDNA screening has precluded the acquisition of chorionic villus sampling and amniocentesis samples. 17 Collecting samples to be used in informative studies is becoming increasingly difficult, as it has been for rare disorder studies in the past. To address the deficit of traditional sample sources used for cfDNA assay validations, we employed a risk-based foundation study supplemented by a targeted case acquisition project. Along with the utilization of remnant control samples, these two groups of pregnancies were combined for analysis. This approach still maintains the essential blinding requirement for validation studies. The study population consisted of two cohorts: a prospectively collected, risk-based group, and prospectively collected cohorts of known affected and control negative cases. (Details of the specific studies can be found at PRO-111 Healthy: https://clinicaltrials.gov/ct2/show/NCT02797743?term=Progenity&rank=4, PRO-101 Samples: https://clinicaltrials.gov/ct2/show/NCT02430584?term=Progenity&rank=6, PRO-100: https://clinicaltrials.gov/ct2/show/NCT02317965?term=Progenity&rank=7) The Laboratory Directors resulting the samples were blinded to all demographic and sourcing information for every sample.
The risk-based cohort, Progenity PRO-100 Obstetrix Study, consisted of a population of pregnant women aged 18–54 at 8–24 weeks’ gestation (inclusive), who had one or more high-risk factors for fetal chromosomal aneuploidy (Tables 1 and 2). All women who elected to undergo an invasive diagnostic procedure for fetal karyotyping were eligible for enrolment. All women in this risk-based cohort provided written informed consent and the study was approved by the Western Institutional Review Board (WIRB #20142299).
Demographic characteristics. Prospective risk-based cohort, Progenity PRO-100 Obstetrix Study (n = 240).

Indications for invasive diagnostic procedure. a Prospective risk-based cohort, Progenity PRO-100 Obstetrix Study.

a103 (43%) of the patients in this cohort had multiple indications, so the total number does not equal 240 and the percentages are >100%.
This approach alone was inadequate to meet the recruitment targets and timelines of the study. The advent of cfDNA screening among high-risk women, and the concomitant reduction in invasive diagnostic procedures, resulted in far slower and less productive recruitment into the PRO-100 study than originally anticipated in 2015, and supplementation of both case and control samples was essential. Additional samples were collected from pregnancies diagnosed with one of the target fetal aneuploidies (Progenity PRO-101 Case Collection Project), and stored without any further evaluation. No additional testing of these samples or exclusion for low fetal fraction or any other reason occurred prior to blinded validation by the Laboratory Directors. These additional maternal blood samples from affected and continuing pregnancies were frozen for later inclusion in the final blinded analysis. This additional source of samples enabled achievement of the original target study sample numbers. Both euploid (Progenity PRO-111) and aneuploid (Progenity PRO-101) samples previously stored at the sponsor’s laboratory without any initial evaluation were included with the prospectively collected samples and analyzed for overall efficacy. The demographic information associated with these samples is summarized in Table 3.
Demographic characteristics. Case Collection Project Progenity PRO-101/111 Study (n = 1195).

All samples, both those collected under the prospective PRO-100 study and the supplemental samples collected and frozen for future analysis, were blinded to testing personnel for identifying characteristics, associated ploidy, and sex chromosome status. Retrieved storage and prospectively collected samples were bar-coded and tabulated in separate password-protected identification spreadsheets kept by the sponsor and the clinical investigation teams. The blinded samples also were randomized across test runs, to avoid any perceived clustering of apparent aneuploid or sex chromosome results. Validation of the Progenity test results with the clinical outcome (karyotype/microarray) was completed after the data were entered, the study closed and the relevant results merged.
Venous blood (approximately 20 mL) was collected from study participants in a cell-free BCT tube (Streck. Omaha, NE, USA). Sample tubes were shipped directly to Progenity, Inc. for processing and storage within five days of collection. Plasma was twice centrifuged, and the supernatant was manually pipetted without disturbing buffy coat. Storage was in 2.2 mL aliquots in cryovials (VWR International. Radnor, PA, USA) at −80°C.
We employed a unique application of MIPs. The key innovation behind this technology is the use of specific primers that anneal to a subset of repeated regions dispersed throughout the genome. Many of the captured sequences are sufficiently distinct to be aligned uniquely to the genome after massively parallel sequencing, requiring fewer reads to achieve equivalent or better performance than most shotgun sequencing cfDNA tests. Although they are members of a dispersed family of genomic sequences, individual members are distinguishable, mapping to and targeting chromosomes of interest (i.e. chromosomes 21, 18, 13, and the sex chromosomes).
Prior to cfDNA extraction at the time of processing, frozen plasma aliquots were thawed overnight at 4°C and centrifuged at 3000g for 5 min to pellet any cryo-precipitate. The plasma supernatants from 96 samples (including controls) were transferred to deep well plates (Arctic White; Bethlehem, PA, USA) for cfDNA isolation using a customized DynaMax cfDNA extraction protocol (Thermo Fisher Scientific; Waltham, MA, USA) adapted for a Microlab Star liquid handling system (Hamilton Robotics; Reno, NV, USA). Isolated cfDNA samples were eluted from the DynaBeads into a single low-bind 96-well polymerase chain reaction (PCR) plate (Eppendorf) for testing.
MIPs, and similar single-molecule oligo-capture technologies, have previously been used in multiplex target-enrichment reactions for human exon and gene resequencing.18–22 The cfDNA test described here uses a novel MIP strategy developed at Progenity, Inc. to enrich and tag specific genomic sequences for next generation sequencing without the need for a highly-multiplexed target capture reaction. The advantages of this approach include reduced assay complexity and high-sequence reproducibility while maintaining a wide-breadth of genomic representation not achievable in targeted cfDNA assays. It also provides a mechanism to interrogate each chromosome utilizing a single probe.
To achieve this, we set out to design a MIP with arm sequences that target thousands of uniquely alignable repeat elements dispersed throughout the human genome (Figure 1). Probe-design software developed at Progenity, Inc. was used to search the human genome for same-strand-targeting probe pairs that met two major design criteria: (1) the probe binding sites repeat across the genome (with no specification of repeat element type) and (2) the probes flank a polymorphic gap sequence that varies from site to site. Under these broad criteria, the probe design software generated >50,000 candidate probe pairs, many of which were unlikely to meet our needs. We filtered for probes most likely to work in a cfDNA assay by adding the following criteria: (1) the major amplicon length of the captured sequences must be <100 bp to allow hybridization to short cfDNA fragments, (2) the unique alignment rate of the amplicons generated must be >60%, (3) the proportion of potential sites with exact match to the primer sequences must be >60%, and (4) >500 sites must be targeted on Chr21. Over 200 candidate probe sequences were designed and compared through an iterative testing process. Top-performing probes were selected for their high overall capture efficiency and ability to generate enough unique Chr21 reads to achieve the desired performance on trisomy 21 samples. We ranked probe performance by calculating the coefficient of variation (CV) of the proportion of aligned reads on Chr21 in healthy cfDNA samples and carried top-performing probes through additional development stages.
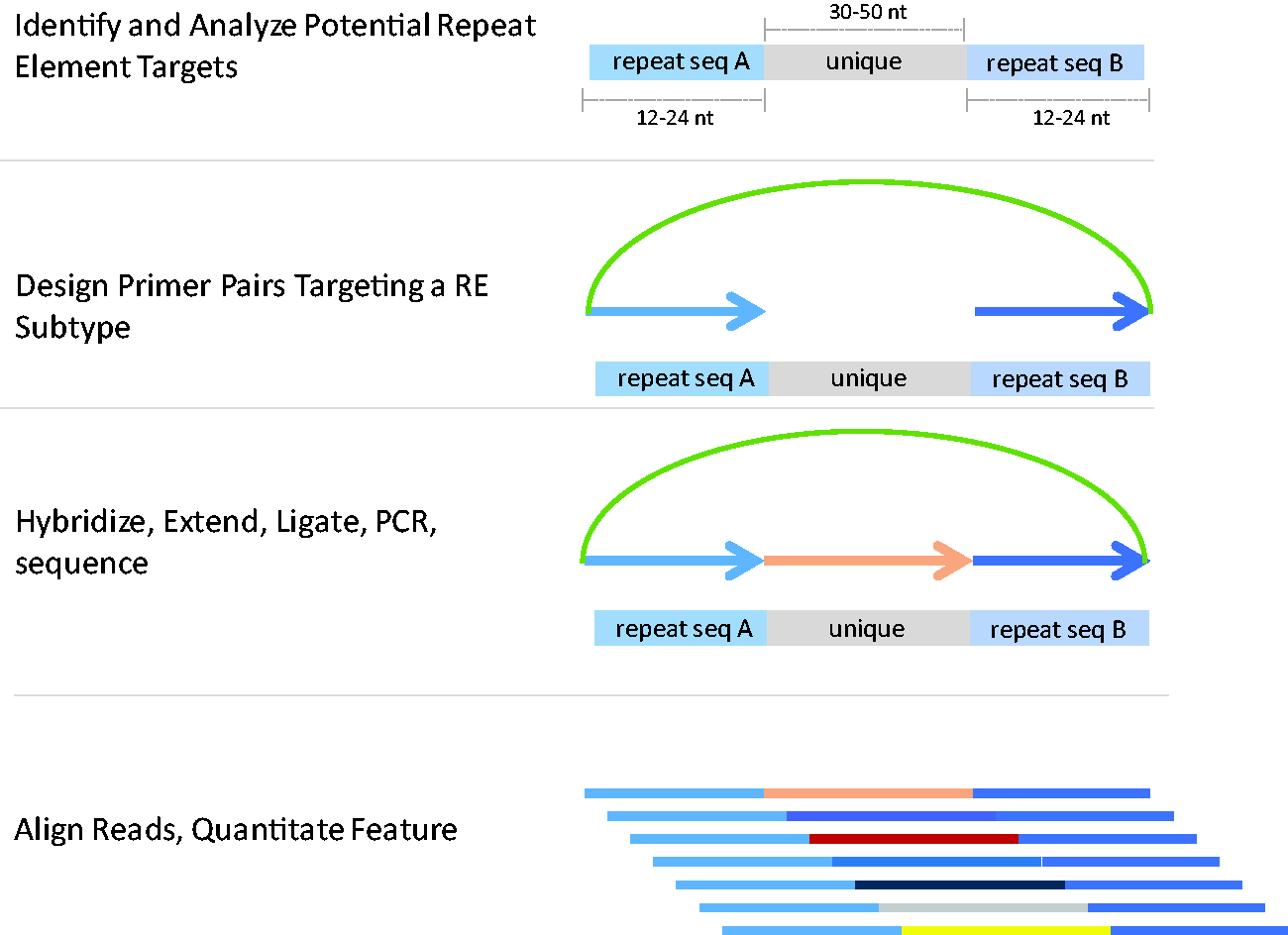
MIP Design for a cfDNA assay. Proprietary software was used to design and filter for probe pairs that can be used to capture repeat elements in the human genome that flank unique or variable sequences (top panel). A backbone containing universal PCR binding sites was added to the selected probe pairs to generate a Molecular Inversion Probe (MIP; 2nd panel). The target capture reaction is composed of three steps – hybridization to the template, polymerase-mediated gap filling, and ligation to generate a single-stranded (ss) circular DNA molecule (3rd panel). The ssDNA is amplified by PCR and the resulting libraries are sequenced. The reads are trimmed, filtered, and aligned. The unique gap sequences allow for accurate alignment of most reads and accurate quantitation of chromosomes (4th panel).
Once a small number of high-performing probes with desirable Chr21 CV (<0.0055) had been identified, additional optimization was performed to maximize the capture reaction for each probe. The final probe selected for the assay was synthesized at scale (Integrated Diagnostic Technologies; Coralville, IA, USA) and tested. The representation of targeted sites scales roughly with the size of each human chromosome (Figure 2). In all, the probe captured ∼200,000 sites across the genome, and >20,000 on Chr21 (Figure 3). For improved quantitation, the capture probe contains a 6 nt-long unique molecular identifier sequence, which allows for the reduction of PCR duplicates and calculation of single capture events from the sequencing reads. 19
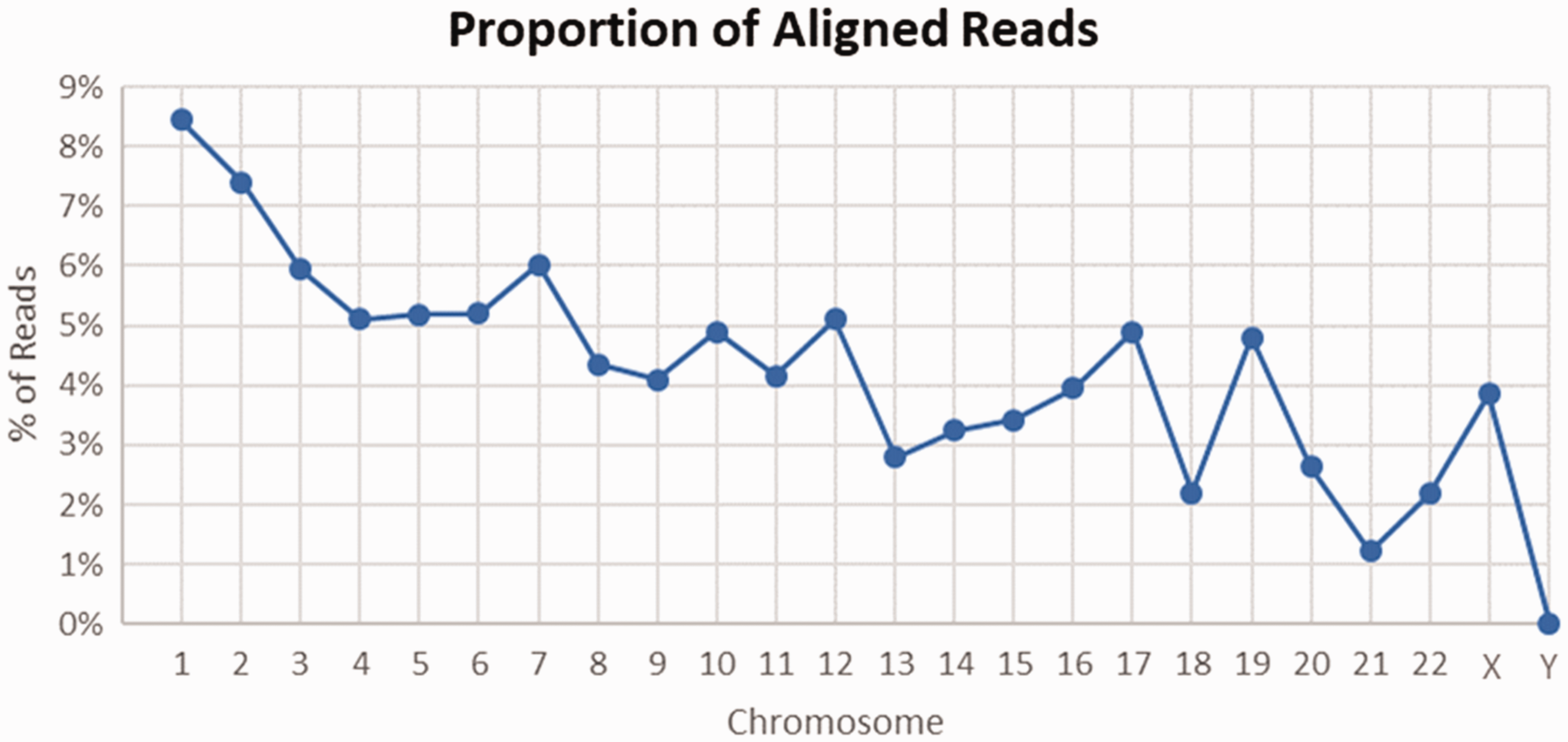
The proportion of reads that align to each chromosome in the cfDNA assay is roughly proportional to the size of the chromosome. The exceptions are chromosomes with high-GC content (e.g. Chr17, Chr19, and Chr22). These chromosomes are likely enriched for the repeat element family targeted by the selected MIP.
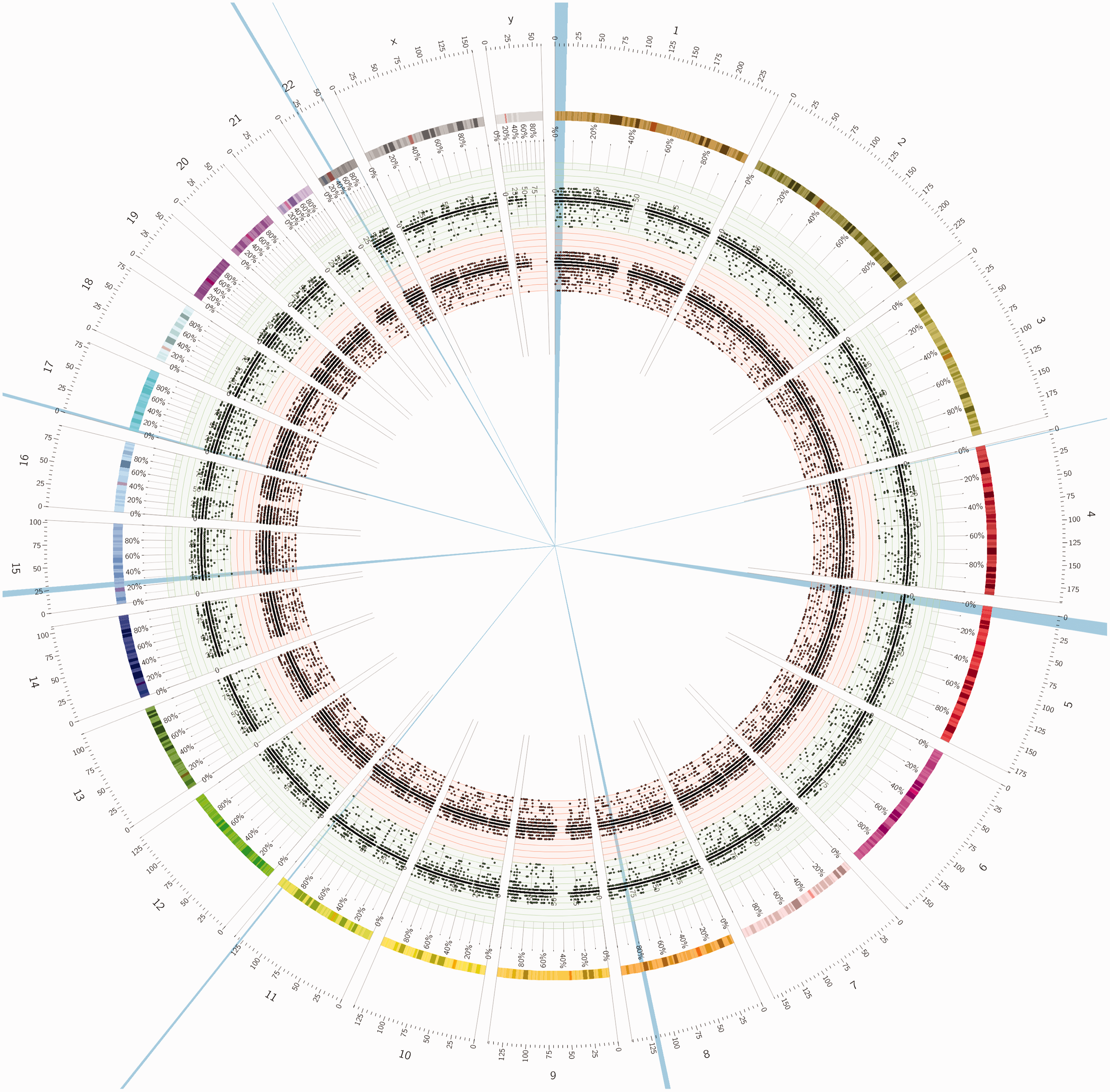
Circos plot indicating the number and location of sites targeted by the assay probe. Chromosome number is indicated around the outer perimeter. The points in the inner ring (orange band, >70,000 points) indicate the sites targeted with no mismatches allowed in the arm sequences. The points in the middle ring (green band, >130,000 points) indicate the sites targeted when 1 mismatch is allowed in the arm sequences. >200,000 sites are targeted in the assay.
Under the MIP cfDNA Assay Protocol, the cfDNA sample is mixed with the identified capture probe and incubated in a thermal cycler to generate hybridized probe-cfDNA product (Figure 1). Standard MIP extension/ligation protocols were modified to capture repeat sequences from cfDNA. The single stranded circular DNA generated from the capture protocol (Figure 1) was used as template in a universal PCR reaction containing primers that bind to the MIP backbone. PCR product libraries were purified with Ampure XP beads (Agencourt AMPure XP, Beckman Coulter; Brea, CA, USA), sample concentrations were normalized to 1 ng/uL, and 276 samples were pooled into a multiplexed sequencing library (including controls). The library was diluted to an appropriate concentration for patterned flow cell clustering on a NovaSeq 6000 (Illumina; San Diego, CA, USA) according to the manufacturer’s instructions. Typical yields from the flow cell resulted in approximately 12 million raw sequencing counts per sample.
Sequencing reads were filtered for cluster clonality and base quality, demultiplexed, and aligned using the Bowtie 2 alignment tool. A ploidy model and aneuploidy calling algorithm was developed at Progenity, Inc. to determine fetal ploidy for the common trisomies and sex chromosomes from sequencing data generated by the assay. This algorithm is based on a likelihood model that fits the count of unique aligned reads at each MIP site in the genome to produce a per-chromosome estimate of ploidy and its standard error. A T-score (Supplemental Appendix A) representing the ratio of the difference between the estimated value and the hypothesized value to the standard error is generated for each chromosome interrogated within each sample. A single nucleotide polymorphism-based fetal fraction estimator also was developed. A preliminary training cohort was evaluated before blinded sample analysis was initiated. There were 503 euploid samples and 42 aneuploid samples (the common trisomies and sex chromosome aneuploidies) in the training cohort. Once the ploidy model was trained using exclusively euploid data and its performance verified, the blinded sample analysis was initiated. Details of the training cohort analysis are available in Supplemental Appendix A.
The study was powered based on a one-sample Sensitivity and Specificity Test (PASS 16.0.1, NCSS Inc., Kaysville, UT, USA). The analysis focused on the trisomy 21 aneuploidy, given that the other aneuploidies were likely to display markedly lower prevalence. Parameters in the test included an estimated 4.8% prevalence of the aneuploidy in the target population, minimum 80% power for both sensitivity and specificity, type I error rate (alpha) of 5%, desired sensitivity of 99.0% (compared with a lower bound of 90.0%), and desired specificity of 99.9% (compared with a lower bound of 95.0%). The analysis indicated that a minimum sample size of 1400, including at least 67 subjects with confirmed trisomy 21, achieves 80% power for the sensitivity determination and 100% power for the specificity determination using a two-sided binomial test. We constructed 2 × 2 contingency tables comparing test outcome for each aneuploidy to clinical outcome. Sensitivity, specificity, positive, and negative predictive values and their associated two-sided 95% confidence intervals (95% CIs) were then calculated from the contingency tables using Wilson’s score method. 23
Results
There were 1435 participants in the study cohort, including 125 samples with confirmed trisomy 21, thereby exceeding the minimum total and T21 sample sizes required to power the study. The study cohort included 1195 from the previously stored samples and 240 from the prospectively collected clinical group. Although the original PRO-100 study protocol called for the evaluation of Trisomy 16 and microdeletion syndromes, there were no cases in the blinded cohort. Other autosomal trisomies were not seen in this study group; further, sub-chromosomal deletions or duplications also were not available for evaluation as they were not seen in these cohorts.
Two samples were damaged in transit; 1433 samples were tested, of which 19 (1.3% of remaining samples) failed quality control during testing, including low fetal fraction (<2% after repeat testing) and quantity insufficient. Five of these 19 samples were from the prospectively collected clinical group (Progenity PRO-100 Obstetrix Study) and 14 from the previously stored samples (Progenity PRO-101/111 Case Collection Project). Accordingly, 1414 samples received a Progenity test result. Among these validated samples, there were 205 aneuploid and 1209 euploid fetuses.
Table 4 shows the results from the common trisomies and sex chromosome aneuploidies. Table 5 summarizes the test results for gender. One male sample was called female, and seven female samples were called male. These discordancies could be due to testing error, clinical reporting error, or biological interference (e.g. mosaicism, vanishing twins, copy number variants). T-score (Supplemental Appendix A) thresholds for euploid, trisomy, and gender calls were empirically established from the T-scores observed in the training set. Sensitivities and specificities were high, as anticipated (>99%, with a single trisomy 21 not identified). Of note are the high positive (99.0% for trisomy 21) and negative (99.2% for trisomy 21) predictive values for these aneuploidies.
Overall summary of Progenity study results. a
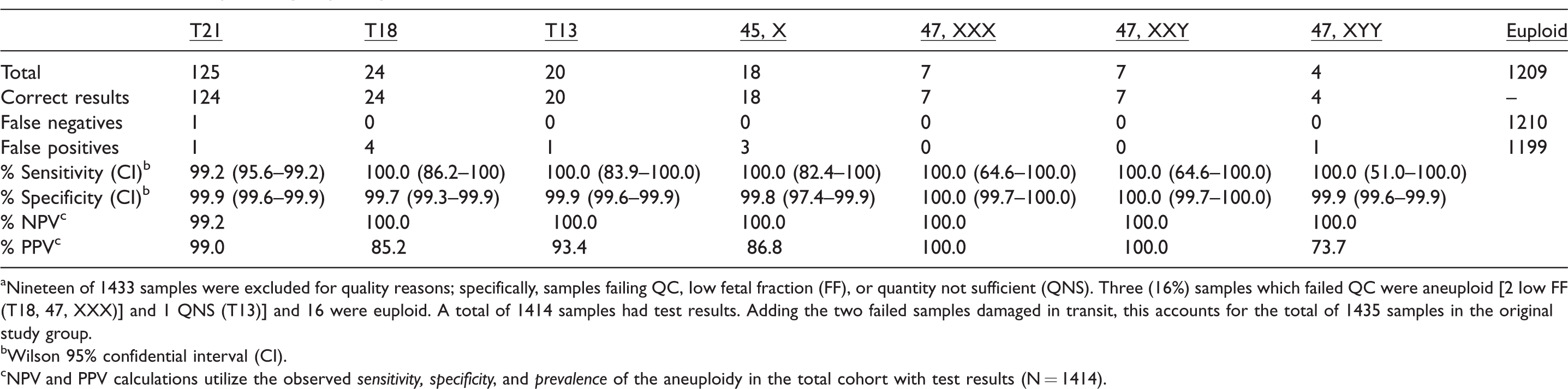
aNineteen of 1433 samples were excluded for quality reasons; specifically, samples failing QC, Iow fetal fraction (FF), or quantity not sufficient (QNS). Three (16%) samples which failed QC were aneuploid [2 Iow FF (T18, 47, XXX)] and 1 QNS (T13)] and 16 were euploid. A total of 1414 samples had test results. Adding the two failed samples damaged in transit, this accounts for the total of 1435 samples in the original study group.
bWilson 95% confidential interval (CI).
cNPV and PPV calculations utilize the observed sensitivity, specificity, and prevalence of the aneuploidy in the total cohort with test results (N = 1414).
Overall Progenity study results for gender.

Total gender error: 8/1414 = 0.6%.
Prevalence rates are in part an explanation of this finding, with a high for trisomy 21 of 8.8% to a low for 47, XYY of 0.3%. Trisomies 18 and 13 had prevalence rates in between, at ∼1.5%. These prevalence rates reflect the study methodology, which included not only high-risk pregnancies, but also the additional stored samples that were enriched with aneuploid fetuses.
The scatterplot distributions for chromosomes 13, 18, 21, and the sex chromosomes are shown in Supplemental Appendix B.
Discussion
In contrast to the current standard of screening for fetal aneuploidy by serum biochemical testing,17,24–26 the level of laboratory performance of cfDNA testing for fetal aneuploidy has become exceptionally accurate, irrespective of the approach or specific laboratory. A recent meta-analysis of 35 relevant publications summarized contemporary test performance. 27 Equivalency at this level of performance is possible as demonstrated by these data.
The novel sequence targeting approach for detecting the major whole chromosome aneuploidies found in this study cohort was validated by the test performance observed. The assay, employing the unique MIPs methodology, demonstrates both sensitivity and specificity of >99% for all the major aneuploidies and sex chromosome abnormalities, as well as comparable positive and negative predictive values reflecting prevalence rates. We speculate that the high sensitivity and specificity of this assay may be a product of the single-probe approach, which theoretically reduces assay susceptibility to batch variation (Figure 4). The performance of this technology is equivalent to other commonly available cfDNA assays. 27
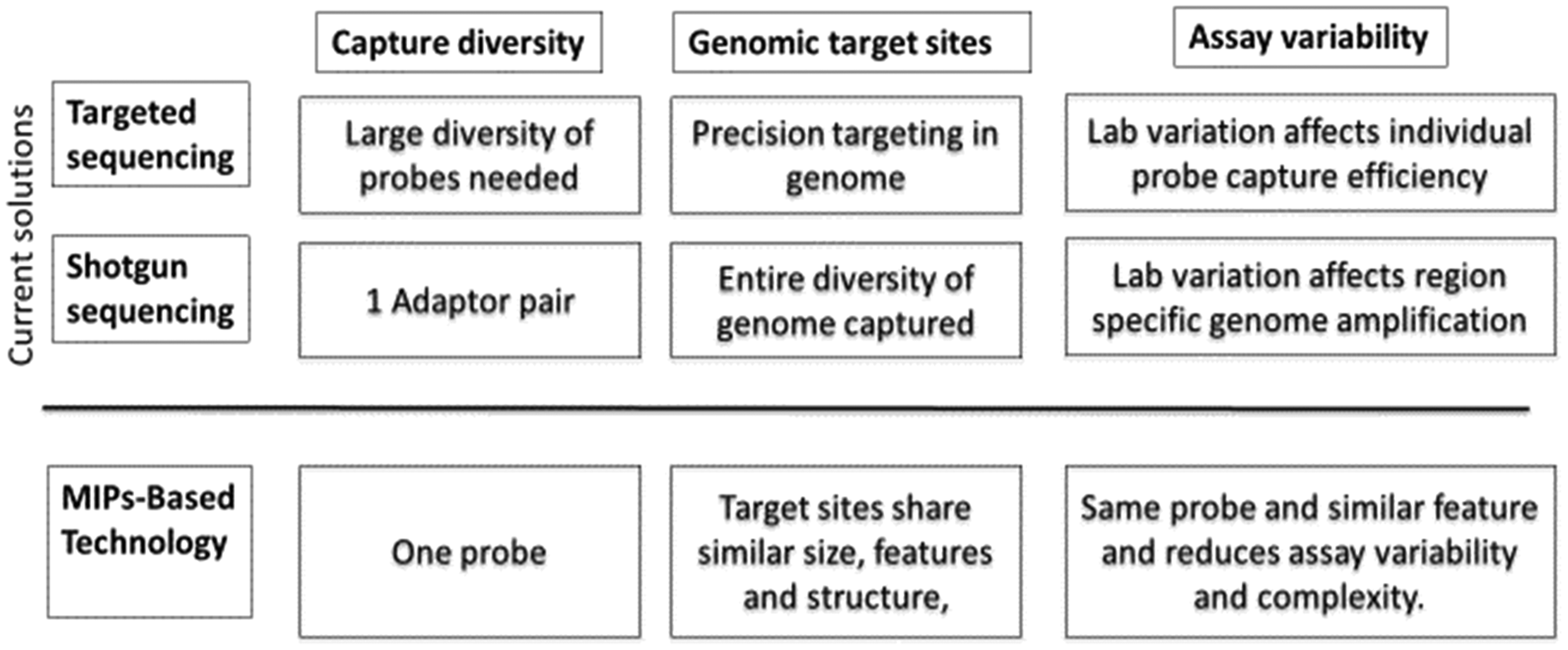
Comparison of sequence technology solutions.
Conclusion
Translating this testing in medical practice will add yet another reason, besides the overall performance, to embrace cfDNA testing as the primary screening for fetal Down syndrome in pregnancy. The impact of cfDNA prenatal screening for chromosomal aneuploidy continues to evolve,28–31 and the introduction of additional innovative sequence targeting techniques as demonstrated in this study will eventually allow test performance to excel, to reach a standard enabling pregnant women at all levels of risk to expect highly predictive screening options.
Supplemental Material
Supplemental Material1 - Supplemental material for Evaluation of a novel screening method for fetal aneuploidy using cell-free DNA in maternal plasma
Supplemental material, Supplemental Material1 for Evaluation of a novel screening method for fetal aneuploidy using cell-free DNA in maternal plasma by Richard P Porreco, Matthew Sekedat, Allan Bombard, Thomas J Garite, Kimberly Maurel, Barbara Marusiak, David Adair, April Bleich, C Andrew Combs, Wayne Kramer, Sherri Longo, Michael Nageotte, Amber Samuel, Jeroen Vanderhoeven, Jeff Buis, Kevin B Jacobs and Jay Stoerker in Journal of Medical Screening
Supplemental Material
Supplemental Material2 - Supplemental material for Evaluation of a novel screening method for fetal aneuploidy using cell-free DNA in maternal plasma
Supplemental material, Supplemental Material2 for Evaluation of a novel screening method for fetal aneuploidy using cell-free DNA in maternal plasma by Richard P Porreco, Matthew Sekedat, Allan Bombard, Thomas J Garite, Kimberly Maurel, Barbara Marusiak, David Adair, April Bleich, C Andrew Combs, Wayne Kramer, Sherri Longo, Michael Nageotte, Amber Samuel, Jeroen Vanderhoeven, Jeff Buis, Kevin B Jacobs and Jay Stoerker in Journal of Medical Screening
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RP, TG, KM, BM, DA, AB, CC, WK, SL, MN, AS, JV: These authors declare no conflicts of interest regarding this study. MS, JB, KJ: These authors declare that they are employees of the study sponsor. AB, JS: These authors declare that they are former employees of the study sponsor.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The funding for the implementation of this study was provided by Progenity, Inc.
Trial registration
Western Institutional Review Board (WIRB), WIRB Protocol Number: 20142299, Approval Date: 9 December 2014, Registration: 9 December 2014, Initial Enrolment: 13 March 2015, Clinical trial ID number: NCT02317965.
Supplemental material
Supplemental material is available for this article online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.