
Abstract
Objectives
To compare the outcomes of sickle cell disease patients diagnosed through neonatal screening with those who were not.
Methods
In an observational multicenter study in Belgium, 167 screened and 93 unscreened sickle cell disease patients were analyzed for a total of 1116 and 958 patient-years of follow-up, respectively. Both groups were compared with propensity score analysis, with patients matched on three covariates (gender, genotype, and central Africa origin). Bonferroni correction was applied for all comparisons.
Results
Kaplan–Meier estimates of survival without bacteremia were significantly higher in the screened group than the unscreened group (94.47%; [95% CI, 88.64–97.36%] versus 83.78% [95% CI, 72.27–90.42%]), p = 0.032. Non-significant differences between both groups were reported for survival without acute chest syndrome, acute anemia, cerebral complication, severe infection, and vaso-occlusive crisis. Significantly lower hospitalization rate and days per 100 patient-years were observed in the screened compared with the unscreened group (0.27 vs. 0.63 and 1.25 vs. 2.82, p = 0.0006 and <0.0001).
Conclusion
These data confirm the benefit of a neonatal screening programme in reducing bacteremia and hospitalization.
Introduction
Sickle cell disease (SCD) is the most common monogenic hemoglobinopathy worldwide, accounting for around 300,000 new cases each year.1,2 In Europe it has received less attention than elsewhere, but with demographic changes, the European incidence of SCD is increasing, and services must be planned accordingly. The introduction of neonatal screening was based on the finding that daily oral prophylactic penicillin from age 4 months gave an 84% risk reduction of Streptococcus pneumoniae bacteremia. 3 Screening programmes include prophylactic penicillin and anti-pneumococcal vaccination, parental education to avoid the most threatening SCD-related adverse events, and prevention of stroke using transcranial Doppler ultrasound. Until now, no randomized clinical trial has been conducted to evaluate the efficacy of neonatal SCD screening. Presumed efficacy is based on the mortality reduction demonstrated, particularly for infants under age 4 years.4,5 The Belgian national neonatal screening programme does not include SCD screening, but local initiatives to implement SCD screening have existed for over a decade, and a database including the diagnosis period (i.e. at birth by neonatal screening or not) and patients outcomes was created in 2008.6,7 This study aimed to compare clinical outcome in two cohorts of SCD patients born in Belgium, the first diagnosed at birth who benefited from the screening programme, and the latter who were unscreened.
Methods
This observational study of two cohorts of patients, diagnosed at birth or later in life, included subjects with severe (HbSS and HbSβ0) and mild (HbSC, HbSD and HbSβ+) genotypes. The Belgian national SCD database was established in October 2008, with hematologists invited to register their patients on a voluntary basis. The registry currently includes patients from six adult and two pediatric centers, of which two are secondary and six are tertiary university care centers. Results published in 2008 8 estimated that about 75–80% of SCD patients living in Belgium were enrolled. Patient data before 2008 were retrieved from medical charts and retrospectively encoded in the database. After 2008, data were prospectively entered in the database for all registered patients and for newly diagnosed subjects. Data were recorded from birth for patients diagnosed through the neonatal screening programme, or from diagnosis following the first contact in the participating center, and until last follow-up visit, or death. Universal neonatal screening was progressively implemented in Brussels from 1994, extending to all Brussels maternity wards by 2000. The screened patients, mostly coming from French speaking Africa, tended to be concentrated in the French speaking part of Belgium, particularly in Brussels and Liège. In 2001, universal neonatal screening was proposed in Wallonia, the French speaking Belgium region. It was performed regardless of patient’s insurance cover, country of origin, or immigration status (legal or not). Since 2003, it includes neonates born under the care of all maternity facilities in Brussels and two other maternity facilities in the Wallonian region.
Umbilical cord blood samples were screened initially using an isoelectric focusing technique (Perkin Elmer Life Sciences, Zaventem, Belgium), and from 2008 using a capillary electrophoresis technique (Sebia Benelux, Vilvoorde, Belgium). Heel prick samples were screened by tandem mass spectrometry. 9 If a hemoglobin variant was detected, further analysis was performed using high-performance liquid chromatography (BioRad, Hercules, California, USA) or DNA analysis. Children presenting with SCD were referred to a specialized center for clinical management. Patients with SCD identified, either through screening or diagnosis following their first SCD-clinical event, were seen and followed regularly in the participating centers according to the policy of each center. All SCD patients received a prescription for recommended oral antibiotic prophylaxis to at least age five. The usual nationally recommended immunization schedule was provided, including conjugate vaccines against Haemophilus influenzae type b (four doses) since 1994, and Streptococcus pneumoniae (three doses) since 2004. From 1999 one center used transcranial doppler ultrasonography for SCD patient follow-up, and this technique is now being extended to all centers. Disease modifying therapies such as Hydroxyurea (HU),10–12 chronic transfusion, and hematopoietic stem cell transplantation (HSCT)13–16 were applied according to each center’s policy, but mostly following the usual indications that have been detailed in previous international publications.17–20 HU was mainly prescribed for patients with recurrent painful episodes and acute chest syndrome (ACS).
For patients lost to follow-up, last available data were from the last visit. After HSCT, only vital status was recorded, and cause of death if applicable. The proportion of patients developing major SCD-related events, the incidence of a first major SCD-related event, hospitalization (number and days), and treatments were analyzed in both groups. Major SCD-related clinical events were defined as: ACS, vaso-occlusive crisis (VOC), acute anemia (Hb < 60 g/L) requiring transfusion, cerebral complication (stroke or transient ischemic attack), severe infection (bacteremia, meningitis, pneumonia, osteomyelitis), and among patients with severe infection, episodes of bacteremia (defined as fever associated with altered general status and identification of a pathogen on blood culture). The most common reasons for hospitalization were acute pain crisis (dactylitis, VOC, ACS), infections, and acute anemia episodes requiring transfusion.
Informed consent was obtained from all patients (or parents/legal guardians). For patients deceased before 2008, data were collected retrospectively (Central Ethics Committee Approval N°: AK/07-11-64/3473). Local ethics committees approved data collection and registry. The dataset included the diagnosis (date, method, and genotype), demographics, SCD-related events, transfusion, hospitalizations, immunization, steady-state biological parameters, radiological data, the presence of chronic organ damage (e.g. cerebral vasculopathy, renal failure), treatments, vital status, and if appropriate, cause of death. For those lost to follow-up, survival data were removed from consideration from the date of the last visit. The study database was locked in December 2012.
Categorical and continuous variables were analysed with Graphpad Prism® software 5.0 (Graphpad Software Inc, San Diego, USA). Kruskal–Wallis test was used to compare continuous data of multiple groups, generating median values. Chi-squared and Fisher’s tests were used to compare data expressed as proportions. The estimate of survival to remain free of specific SCD-related complications was generated for the time to first event in the screened versus the unscreened group. Kaplan–Meier and the 95% confidence interval (95% CI) were calculated for all SCD-related complications. Survival curves of the different events were compared by the log-rank test. A p-value less than 0.05 was considered statistically significant. Propensity score analysis was performed to match patients between groups using three covariates: gender, genotype, and central Africa origin. The covariate balancing propensity score R package was used to perform the propensity score, estimating an average group effect and requiring an exact match, using covariate balancing, which has been showed to be superior to traditional logistic regression approaches and boosted classification and regression trees. 21 An absolute standardized difference less than 10–15% was considered to support the assumption of balance between the groups because it is not affected by the sample size, unlike p-values, and it may be used to compare the relative balance of variables measured in different units. 22 The percentages of categorical variables before and after propensity score are presented. For time-to-event analyses, application of propensity scores using inverse probability weighting (IPW), rather than matching, stratification, or adjustment, were used as it produces effect estimates with minimal bias.23,24 For binary outcomes, the effect of neonatal screening method on the outcome is estimated by the regression of the outcome on an indicator variable denoting neonatal screening method assignment and the propensity score (inverse probability of neonatal screening weighting). For continuous variables, the effect of the neonatal screening method was estimated by the linear regression of the outcome on an indicator variable denoting group assignment; the covariates present in the propensity score (inverse probability of neonatal screening weighting). Bonferroni correction was applied in all analyses. A p-value less than 0.0083 (0.05/6) was considered statistically significant as six variables were tested.
Results
Among the 469 patients recorded in the registry, 260 were born in Belgium, including 167 screened at birth and 93 not screened, accounting for 1116 and 958 patient-years of follow-up, respectively (Figure 1). Demographic and follow-up characteristics of both cohorts are summarized in Table 1. As reported previously,
7
in the screened cohort six patients were lost to follow-up and two patients died (age 1.5 and 2.1 years) during the 1990, before the complete programme was implemented. In the unscreened cohort, three patients were lost to follow-up and one died (age 18.7 years; hemorrhagic stroke). Death rate was similar in both groups, i.e. 1.2% (0.18 per 100 patient-years) and 1.1% (0.10 per 100 patient-years), respectively (p = 1). Screened patients were statistically younger at diagnosis and at last follow-up. They also had a shorter period of follow-up. Most of the patients have a severe genotype and the majority of the families originate from Central African countries (70% from the screened and 77% from the unscreened cohort), and particularly from the Democratic Republic of Congo (60%). The unscreened group had significantly higher rate and days of hospitalization than the screened group.
Flow chart of the SCD patients born in Belgium. Demographic and follow-up characteristics of the Belgian cohort of sickle cell disease patients.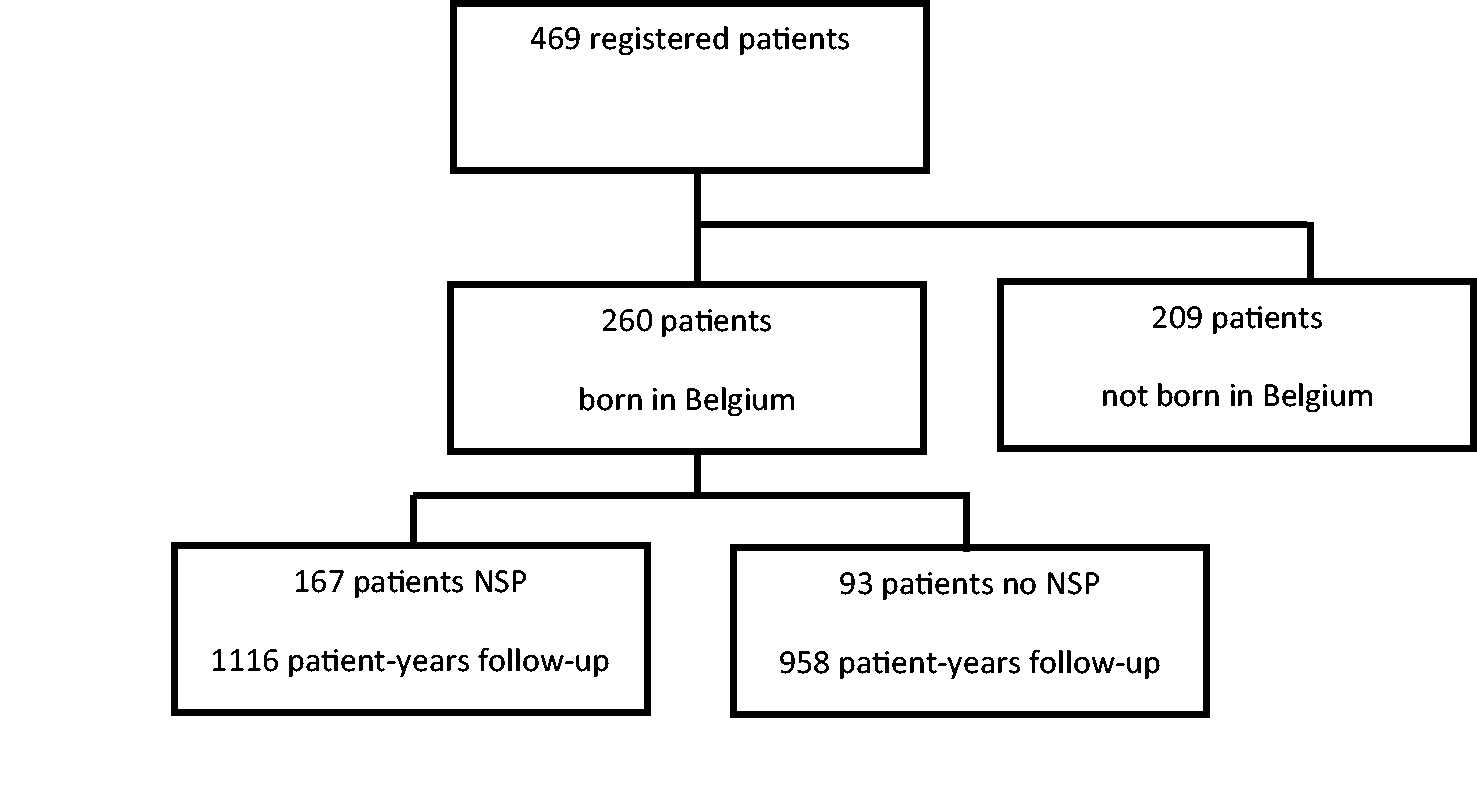
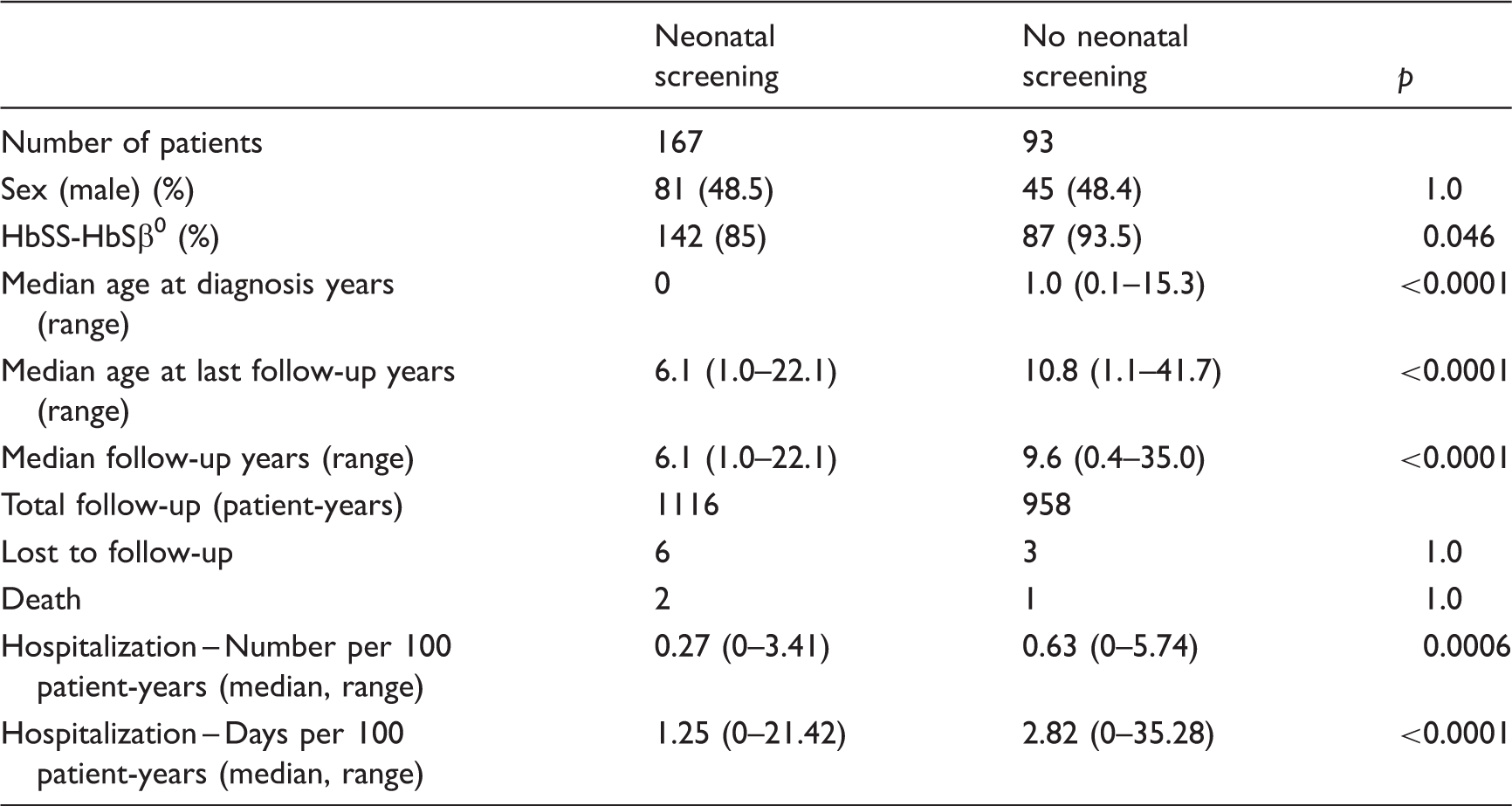
Patient characteristics by neonatal screening status for propensity-matched patients.
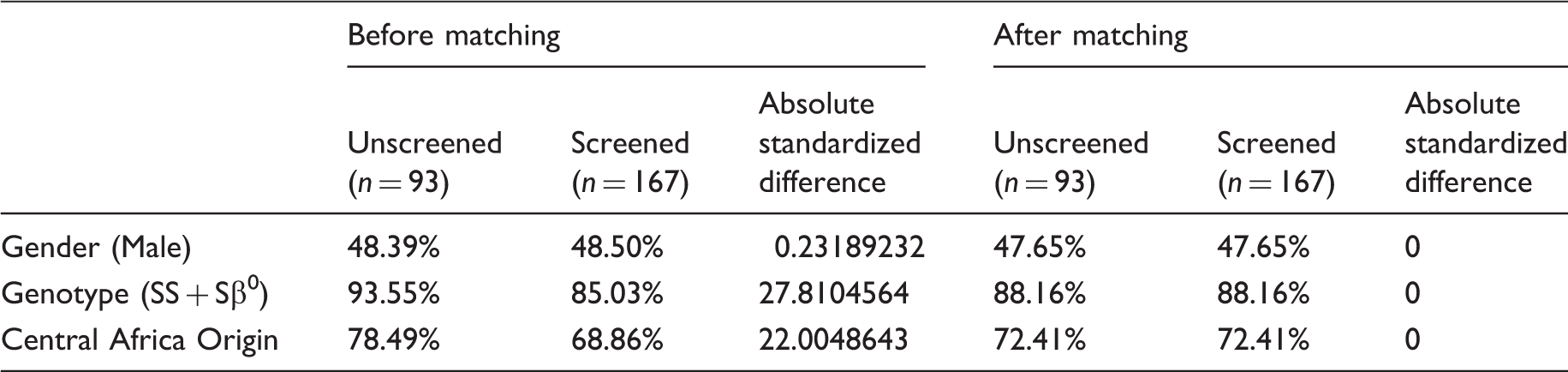
Differences between the three chosen covariates included in the propensity score disappeared after covariate balancing propensity score was run because all absolute standardized differences are equal to 0. Moreover, all patients remain in the sample after propensity.
Age of first event from diagnosis for propensity-matched patients.
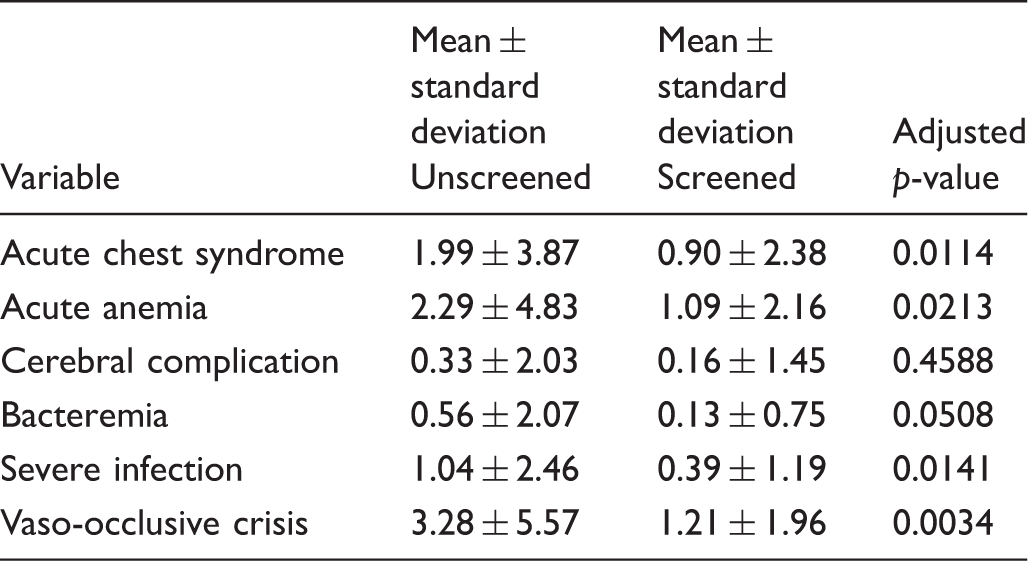
Weighted means and standard deviations by group, and adjusted p-value of the group effect from each linear regression. Bonferroni correction was used to reach the significant level. As there were six comparisons, a p-value less than 0.0083 (0.05/6) was considered statistically significant.
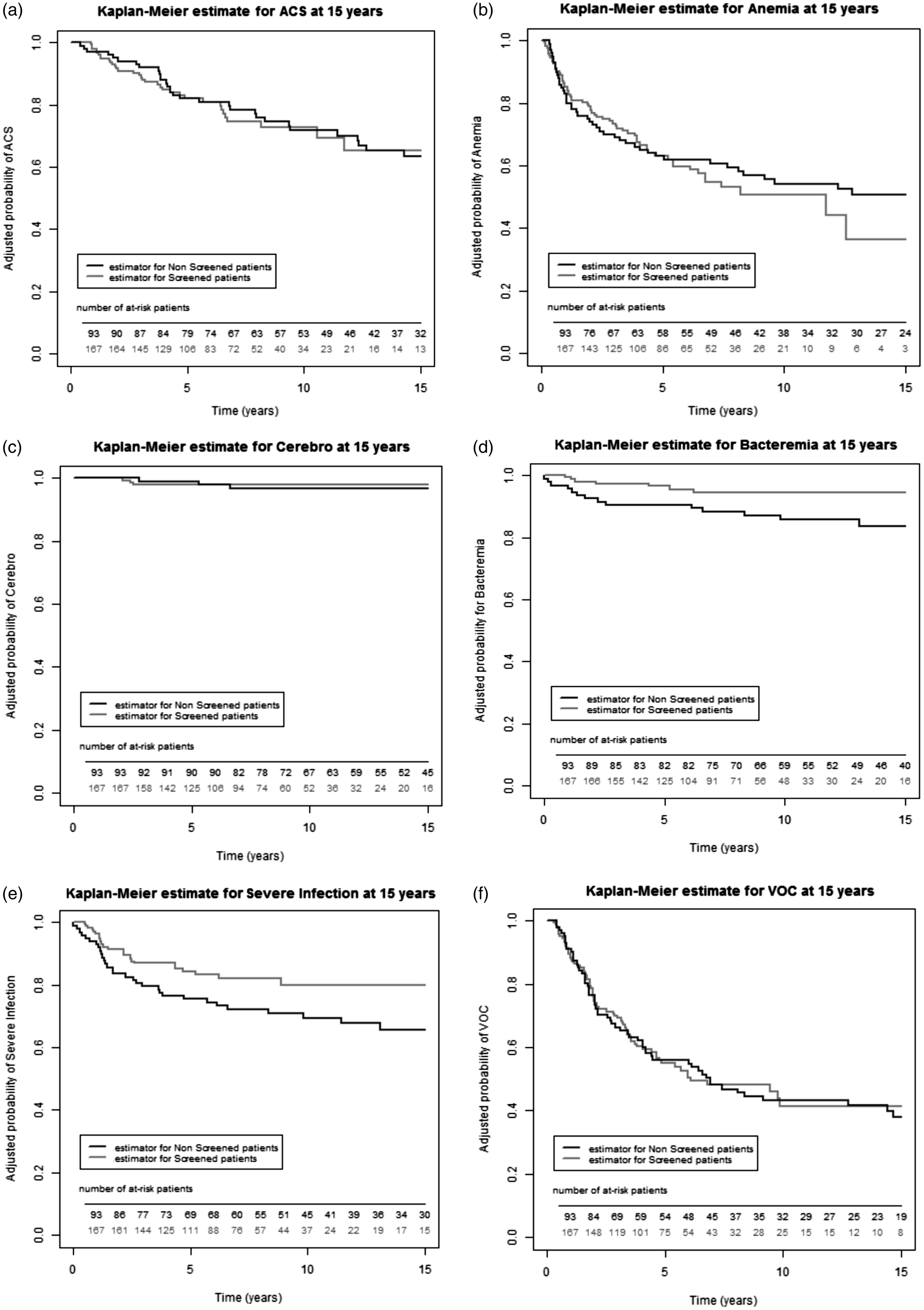
(a) 15-year Kaplan–Meier estimate without acute chest syndrome were not different in both groups (65.17% [95% CI, 51.07–76.13%] vs. 63.57%; 95% CI, 51.03–73.71%], p = 0.821). (b) 15-year Kaplan–Meier estimate without anemia were not different in both groups (36.42%; [95% CI, 19.15–53.95%] vs. 50.66%; 95% CI, 38.72–61.42%], p = 0.586). (c) 15-year Kaplan–Meier estimate without cerebral complication were not different in both groups (97.98%; [95% CI, 72.27–99.33%] vs. 96.80%; [95% CI, 89.73–99.03%], p = 0.755). (d) 15-year Kaplan–Meier estimate without bacteremia was significantly higher in the screened group than the unscreened group (94.47% [95% CI, 88.64–97.36%] vs. 83.78% [95% CI, 72.27–90.42%], p = 0.032). (e) 15-year Kaplan–Meier estimate without severe infection were not different in both groups (80.00% [95% CI, 71.21–86.36%] vs. 65.68% [95% CI, 53.64–75.30%], p = 0.054). (f) 15-year Kaplan–Meier estimate without VOC were not different in both groups (41.38% [95% CI, 30.87–51.56% vs. 37.90%; [95% CI, 26.50–49.23%, p = 0.953).
Therapeutic intensification was applied to 43% (72/167) of those in the screened group and 83% (77/93) of those in the unscreened group. The median ages at therapeutic intensification were 3.2 years (0.8–12.4 years) and 5.1 years (0.7–31.9 years), respectively. HU treatment was more frequently prescribed in the unscreened group than the screened group (45% vs. 30%; p = 0.015), but introduced at a younger age in the screened cohort: 2.6 years (range: 0.6–7.9 years) versus 4.5 years (range: 0.7–31.9 years) (p = 0.0001). This is still the case when considering patients with severe genotype. HSCT was performed in 10% of the screened and 30% of unscreened patients (p = 0.0001). Median age at transplantation was 4.9 years (range: 1.1–12.4 years) and 4.55 years (range: 1.1–15.3 years), respectively (p = 1). All the HSCT patients were homozygous for Hb S.
Discussion
Data from Belgium and elsewhere show that, even in developed countries, SCD patients have higher morbidity and mortality rates than the general population.5,25–29 Our data indicate that bacteremia is less frequent in screened patients, possibly because of systematic antibiotic use, widespread immunization with conjugated vaccines against Streptococcus pneumoniae and Haemophilus influenzae type b,3,30,31 or parental education from the time of diagnosis.25,26 This underlines the importance of comprehensive medical follow-up associated with universal neonatal screening.25,32 A dedicated nurse is probably also crucial, to provide information, discuss treatment, and reinforce contacts with SCD patients and their families. They also organize annual exams and are a key point of contact with social workers and educational teams, work crucial to improve adherence to treatment and empowerment of families. Adherence to antibiotic prophylaxis might be less good in the unscreened cohort because education starts later, but the efficiency of all other implemented measures could explain why, while the bacteremia rate is increased, the survival rate is not affected. The lost to follow-up rate (3.6%, 6/167) in our neonatal screening programme was similar to that from the London and Dallas cohorts.26,28 Our screening programme death rate (1.2% (2/167); 0.18 per 100 patient-years) was similar to the London cohort, but inferior to the rate in the Californian, Dallas, and Brazilian cohorts.25,28,29 Our more favorable results might be explained by specific programmes developed in participating centers, easy access to medical care for all patients across the country, and the effort expended on education of patients and families over many years.
Our unscreened cohort is older and has a longer duration of follow-up, which might explain why those patients developed more complications than the screened cohort. Nevertheless, Kaplan–Meier analysis on the entire cohort confirmed the clear advantage of the screened group in the risk of developing bacteremia. A lower hospitalization rate and days were also found in the screened group. These advantages may be explained by several factors. Screened patients and their families may be more concerned by the disease and more prompt to react to clinical signs due to educational and comprehensive care efforts implemented earlier. The effective implementation of antibacterial prophylaxis may also contribute, as may rapid implementation of HU in screened patients as indicated in recently published guidelines. Importantly, fatalities or bacteremia that occur before the diagnosis of SCD were not recorded in our database due to our methodology, and this might lead to underestimation of the rate of these complications in the unscreened cohort. Our results clearly demonstrate the long-term positive impact of neonatal screening on the risk of developing bacteremia, but screened patients have similar adjusted 15-year overall survival and survival without ACS, acute anemia cerebrovascular disease, and severe infections to the unscreened cohort. The reason for the lower hospitalization rate in screened patients is not clear. Earlier introduction of HU might be an explanation, with less complicated ACS or VOC, but these data were not systematically recorded in the registry as we did not record the number of specific events once the first had occurred. Our methodology does not allow us to compare the incidence of VOC or ACS over time, expressed as the number of events per 100 patient-years.
This study has several limitations, including the relatively small size of our cohort, and the possibility of incomplete data in the retrospective part of the study. In addition, events such as fatal sepsis, VOC, or ACS occurring before adequate diagnosis in the unscreened cohort are misdiagnosed and missing in the records. The morbidity data are quite convincing in terms of advantages, with significantly lower bacteremia and lower hospitalization rate and days per 100 patient-years in the screened patients, so even though our data showed no difference in overall survival, the better event-free survival for bacteremia is important. Indeed, those infections are potentially associated with a higher co-morbidity (VOC, ACS, severe anemia requiring transfusion).
Conclusion
In Belgium, neither antenatal nor neonatal screening have been organized or funded nationally, 33 but our results suggest that universal neonatal screening is feasible and effective to detect SCD and improve SCD patient care in Belgium, and that children diagnosed with SCD benefit from comprehensive expert medical care in dedicated centers. This includes education of patients and parents on complication prevention, lifestyle, and management of fever and pain. It requires collaboration among parents and patients, doctors, nurses and social workers, not only in case of acute events, but also for prevention, early detection, and treatment of chronic organ damage, to enable patients to better control their disease. Integrated care allows earlier introduction of disease modifying therapies to reduce morbidity and improve quality of life, as our data and recent French data 32 both show. SCD screening, either before conception or during the antenatal/neonatal period, should be extended to the whole country.
Footnotes
Acknowledgement
The authors thank Dr Eric Sariban for critically reviewing this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fonds IRIS Recherche Brussels, Belgium (2007) and an Educational Grant kindly provided from Novartis Pharma Belgium.