
Abstract
Objective
To determine the prevalence of α-thalassaemia in β-thalassaemia individuals in a Chinese population.
Methods
The standard diagnostic marker for β-thalassaemia was elevation of the Hb A2 level (>3.5%) with low mean corpuscular volume. The common α-thalassaemia mutations were studied by molecular analysis in all identified β-thalassaemia carriers.
Results
A prevalence rate of 3.3% for β-thalassaemia was found in our population; α- and β-thalassaemia interactions were found to co-exist in 17.8% of the β-thalassaemia carriers. The -SEA deletion was the most common α-thalassaemia mutation co-inherited with β-thalassaemia, followed by the -α3.7 deletion, the -α4.2 deletion, Hb Quong Sze, and Hb Constant Spring.
Conclusion
Our results suggest that it could be valuable to study co-existing α-globin mutations in subjects with β-thalassaemia trait in a prenatal screening programme, especially in populations with a high prevalence of haemoglobinopathies.
Introduction
In Guangdong province, southern China, the prevalence of β-thalassaemia is around 2.5%, and that of α-thalassaemia is 8.5%. 1 Co-inheritance of these two types of thalassaemia is not uncommon and is a challenge for the screening programme.
The characteristic haematological features of α-thalassaemia are low mean corpuscular volume (MCV), low mean corpuscular haemoglobin (MCH), and normal or reduced haemoglobin (Hb) A2. In β-thalassaemia, low haematological indices in heterozygotes are accompanied by increased Hb A2 (>3.5%). Double heterozygotes for α- and β-thalassaemia are usually diagnosed as pure β-thalassaemia carriers, and α-thalassaemia is generally ignored.2–4
The two thalassaemias vary considerably in their clinical presentations, especially when they exist together. For example, reduction of α-globin synthesis in β-thalassaemia restores globin balance and individuals demonstrate an improved phenotype. 5 Similarly, the coexistence of β-thalassaemia is a background modifier for ameliorating disease severity in patients with haemoglobin H disease (–/−α). 6 In order to design an appropriate screening strategy to detect complex carriers and provide accurate genetic counselling, it would be valuable to document the molecular characterization of thalassaemias, especially the co-inheritance of α- and β-globin gene defects, in a region with high prevalence of thalassaemias.
In this study, we prospectively evaluated the prevalence of co-inheritance of α-thalassaemia in Chinese β-thalassaemia carriers in a large prenatal screening programme.
Methods
Between January 2012 and October 2013, a hospital-based programme for prenatal screening for α- and β-thalassaemias was carried out at a local suburban obstetric unit in Guangzhou region, Guangdong province. The subjects were all indigenous inhabitants, selected from couples who were screened for thalassaemia trait at their first presentation for prenatal care. Both partners were given an ordinary blood test (complete blood count). If both partners had a normal MCV value, no further assessment was undertaken. When at least one partner of a couple had a reduced MCV value, both partners undergo detailed screening for a thalassaemia trait. A pregnancy in which both partners had the same type of thalassaemia was considered to be at risk, and genetic counselling services and prenatal diagnoses were offered.
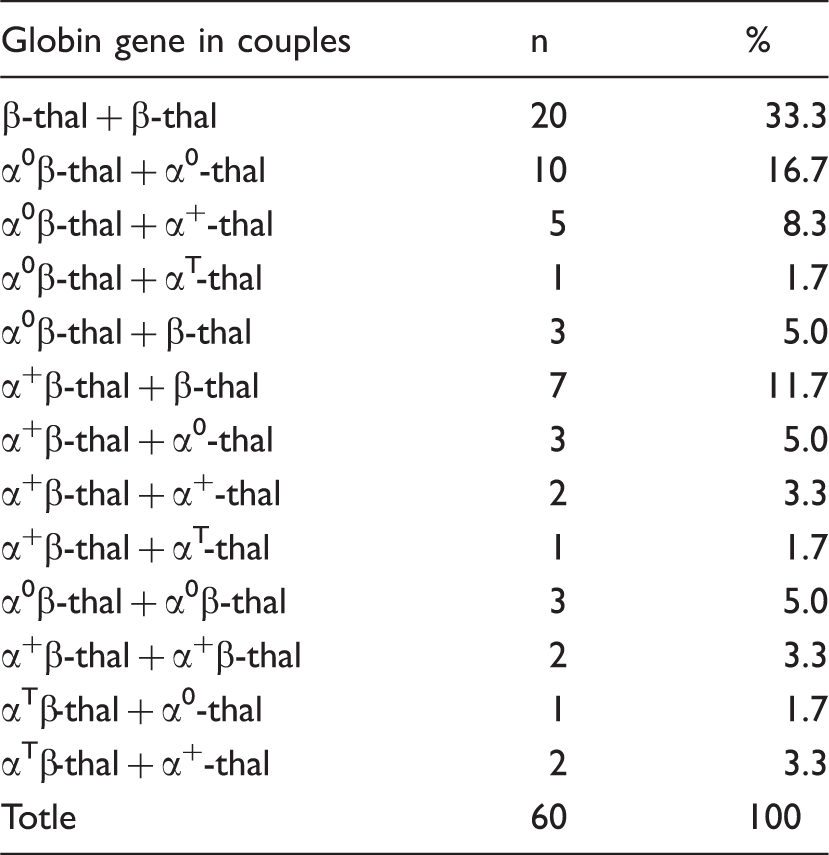
An initial MCV cut-off value of 80 fL was applied. Full blood counts and erythrocyte indices on all blood samples were determined by the Sysmex XE-2100 automated blood cell counter (Sysmex, Kobe, Japan) within 12 hours of venesection. Elevated Hb A2 level (>3.5%), the standard diagnostic marker for β-thalassaemia was determined by a Sebia electrophoresis system, the Capillarys2 (Sebia, Lisses, France). The diagnostic marker for α-thalassaemia is a low MCV value, ruling out the possibility of iron deficiency. Confirmatory laboratory test for α-thalassaemia usually requires molecular diagnosis. Gap-polymerase chain reaction (gap-PCR) is used to determine the three common Chinese α-globin gene deletions (−α3.7, −α4.2, and –SEA). The PCR-reverse dot-blot (RDB) method was used to detect the two common nondeletonal α-thalassaemias (Hb Constant Spring and Hb Quong Sze). Another PCR-RDB assay was used to define 17 known Chinese β-thalassaemia mutations [codons 41–42 (-TCTT), IVS-2-654 (C→T), −28 (A→G), codons 71–72 (+A), codon 17 (A→T), codon 26 (G→A), codon 31 (-C), codons 27–28 (+C), IVS-1-1 (G→T), codon 43 (G→T), −32 (C→A), ATG→AGG, −30 (T→C), codons 14–15 (+G), CAP + 1 (A→C), −29 (A→G) and IVS-1–5 (G→C)]. DNA sequencing was used to determine the unknown α- or β-thalassaemia point mutations.
Results
During the study period, 11,056 couples underwent prenatal thalassaemia screening. Among these, 8750 couples were designated screen negative, and in 2306 couples at least one partner had a positive screening test.
The β-globin gene mutations and their frequencies in 725 individuals with β-thalassaemia.
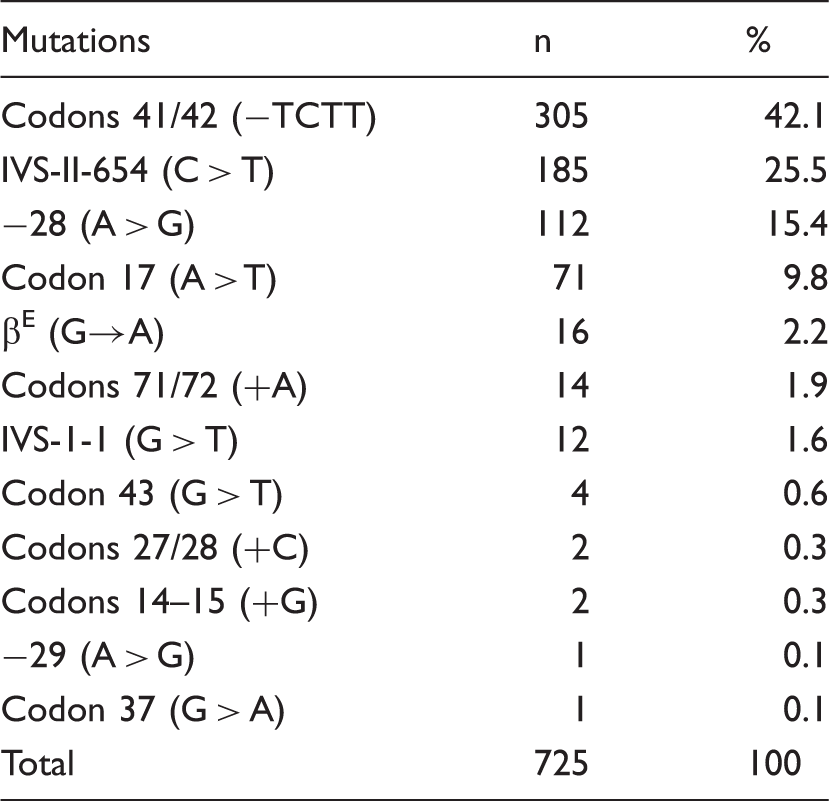
Distribution of α-thalassaemia in the β-thalassaemia carriers.
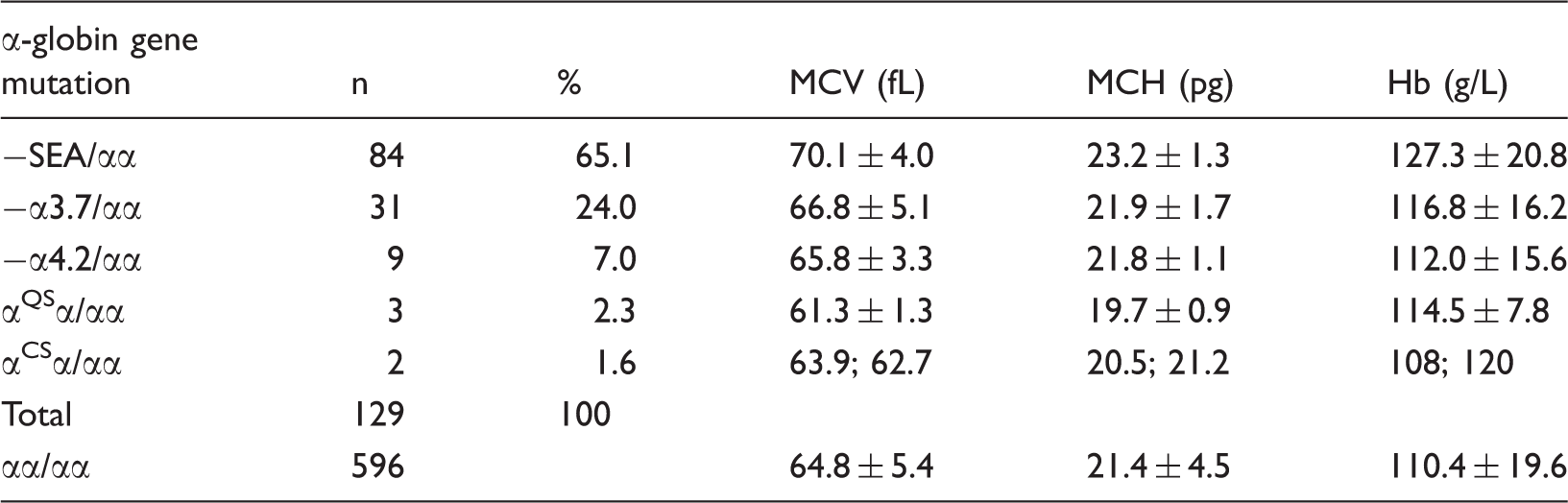
Co-inheritance of α- and β-thalassaemia in 60 couples.
Discussion
Thalassaemia is of major public health importance for Guangdong province, and prenatal diagnosis is the most effective means of reducing the burden of suffering associated with this disorder. China has not yet implemented a population-based screening programme, because of the relatively high cost, and shortage of skilled technologists. A hospital-based screening and preventive programme during pregnancy is the main strategy used. 7 All couples are screened for thalassaemia trait at the mother’s first prenatal visit, to identify the couples at risk and to detect severe forms of thalassaemia in the fetus. In our study we focused only on the co-inheritance of α- and β-thalassaemia.
Conventional screening methods cannot detect minimal changes in the haematological phenotypes occurring in double α-/β-thalassaemia heterozygotes, and misdiagnosis of these double heterozygotes as pure β-thalassaemia heterozygotes may easily occur. DNA analysis of the α-thalassaemia gene in β-thalassaemia carriers is usually needed to differentiate the two types. Co-existing α-/β-thalassaemia is relatively common in Southeast Asia, where 4–20% of the population have α-thalassaemia.8–10 We found a prevalence of 17.8% of α-thalassaemia in β-thalassaemia carriers, which was higher than the general prevalence (8.1%) in Guangdong. Our results suggest that one in every six β-thalassaemia carriers co-inherits α-thalassaemia. This is particularly important in couples who appear to carry discordant forms of thalassaemia according to routine haematologic testing. In the 25 such couples in our study, there were 10 pregnancies at risk for haemoglobin Bart’s hydrops foetalis, 8 pregnancies at risk for deletional Hb H disease, and 2 pregnancies at risk for nondeletional Hb H disease.
In a previous study, we reported finding of the -SEA deletion in 4.4% of β-thalassaemia carriers in urban dwellers of Guangzhou, the capital region of Guangdong province. 11 A much higher prevalence (11.6%; 84/725) of α0-thalassemia was found in β-thalassaemia carriers in the current study, largely because of the different racial and indigenous groups in the two studies. Unlike the city dwellers of Guangzhou which comprise diverse groups coming from across the country, the indigenous residents in the present study are a relatively homogenous group. They live in the suburban area of Guangzhou region, and are mainly farmers, with little progress in their social economic status. This suggests that thalassaemia mutations are not only specific to different populations, but can also differ in frequency between indigenous groups within the same population.
In the past, deletional α-thalassaemia was the focus of screening and we excluded the –SEA deletion in β-thalassaemia carriers. Only couples with both partners being carriers of α0-thalassaemia deletion were offered prenatal diagnoses. The algorithm for identifying pregnancies at risk for thalassaemia has recently changed, 12 and an investigation for common non-deletional α-thalassaemia for detection of couples at risk for α-thalassaemia is included in our prenatal screening programme.
Although the deletional forms of Hb H disease are not fatal, individuals with non-deletional Hb H disease have a more severe phenotype, with earlier presentation, more severe anaemia, jaundice, bone changes, and greater hepatosplenomegaly. 13 Individuals with more severe haematologic phenotypes may require transfusions more frequently than those with deletion Hb H. Some develop transfusion-dependent anaemia after birth,14,15 or suffer from severe anaemia and hypoxia in utero, presenting as Hb H hydrops fetalis syndrome. 16 Because Hb Constant Spring and Hb Quong Sze account for the majority of alleles causing non-deletional Hb H disease in Chinese, only these two alleles are targeted in clinical practice. We recently reported that almost all of Hb Constant Spring carriers could efficiently be detected using a capillary electrophoresis method. 17 However, Hb CS trait could not be identified using this method when it is combined with β-thalassaemia. 18 Hb Quong Sze variant is extremely unstable and degrades rapidly, and is undetectable by routine Hb electrophoresis. The PCR-reverse dot-blot method was therefore used to detect simultaneously the Hb Constant Spring and Hb Quong Sze mutations in β-thalassaemia carriers.
In couples where both partners had β-thalassaemia trait, the detection of α-thalassaemia has clinical significance. Homozygotes or compound heterozygotes for β-thalassaemia who co-inherit α-thalassaemia will have less redundant α globin, and tend to have a less severe condition. This α-/β-thalassaemia interaction alone provides the basis for considerable clinical heterogeneity; the degree of amelioration depends on the severity of the β thalassaemia alleles and the number of functional α globin genes. Co-inheritance of two α-globin gene deletions (usually in the form of −α/−α) or a non-deletional α2-globin gene mutation (αTα/αα) in β0-thalassaemia homozygotes is more likely to produce the clinical phenotype of thalassaemia intermedia, whereas the co-inheritance of a single α-globin gene deletion in the same group of patients is usually associated with thalassaemia major phenotype.19–22 The SEA deletion (−/αα) ameliorates the clinical phenotype of β0/β+ but not necessarily that of β0/β0-thalassaemia. 23 In homozygosity or compound heterozygosity for β+-thalassaemia, co-inheritance of a single α-globin gene deletion is sufficient to produce an ameliorating effect. 24 At one extreme, a patient who had co-inherited Hb H disease with β0/β+-thalassaemia presented clinical manifestations as a β-thalassaemia heterozygote. 25
Just as co-inheritance of α-thalassaemia can reduce the clinical severity of homozygous β-thalassaemia, the presence of β-thalassaemia can also ameliorate non-deletional Hb H disease.26–28 This is important for couples where one partner co-inherits α- and β-thalassaemia, and the other inherits only the α-thalassaemia mutation.
Conclusion
A high prevalence of α-thalassaemia was found in β-thalassaemia carriers. Individuals who co-inherit α- and β-thalassaemia have impaired synthesis of both chains, displaying a markedly improved phenotype. This could be indirectly supported by our findings of significant improvement in MCV and MCH values for β-thalassaemia mutation co-inherited with one or two α-globin gene deletions. Because of a high prevalence of α-thalassaemia in the Chinese population, it is sensible to look for α-thalassaemia in β-thalassaemia carriers within the prenatal screening programme. This has two clinical implications: first, molecular analysis must be used for accurate diagnosis of double heterozygotes in couples presumed to be discordant for α- and β-thalassaemia on haematologic testing. The diagnosis of this double heterozygote state is important because, as opposed to typical β-thalassaemia carriers, these individuals will be at risk of having offspring with severe α-thalassaemia if their partner is also an α-thalassaemia heterozygote. Second, the degree of globin chain imbalance strongly influences the level of ineffective erythropoiesis, and subsequently the overall expression of clinical severity. Reduction of globin chain imbalance through the interaction of α- and β-thalassaemia is expected to modify classical phenotypes of α- and β-thalassaemia. This information should be incorporated into the genetic counselling of such couples.
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Funding
Funding from the Guangzhou Health Bureau (20121A021012, 20131A011066) is gratefully acknowledged.