
Abstract
Thermal and rheological properties of polyamide 6/layered double hydroxide (PA6/LDH) composites were studied. Pristine (U-LDH) and organically modified (M-LDH) clays were used in this study. Some evidence of intercalation was observed on the microstructure of PA6/M-LDH samples, while PA6/U-LDH was characterised by microcomposite morphology as shown by the transmission electron microscopy results. The scanning electron microscopy results showed a complete delamination of the M-LDH in the PA6 matrix while U-LDH was evenly dispersed as immiscible tactoids. In the melt state, the M-LDH had a significant influence on the melt microstructure of PA6 matrix when compared to U-LDH. Differential scanning calorimetry results, both dynamic and isothermal experiments, showed that LDH had a heterogeneous nucleating effect on the PA6 matrix, with the U-LDH showing better nucleating effect when compared to M-LDH. The thermogravimetric analysis results showed that M-LDH had a negative influence on the thermal stability of the composites, while U-LDH improved their thermal stability. The X-ray diffraction and dynamic mechanical analysis results showed that the presence of M-LDH promoted the formation of γ-crystallites in the PA6 matrix, while U-LDH composites were dominated by the α-crystallites. This phenomenon had a positive correlation with increasing content of both LDH clays. An overall improved dynamic mechanical properties were observed for PA6/U-LDH when compared to PA6/M-LDH composites.
Keywords
Introduction
The past two decades have witnessed an exponential growth in the interest on layered double hydroxide (LDH) clays, practically from all fields of science for academic research and industrial application purposes. Owing to the versatility of the LDH clays, that is, flexibility, ease of synthesis, customizability of the chemical composition, potential recyclability, excellent particle size distribution and ability to host a wide variety of organic and inorganic anions within its galleries, they have become the materials of interest. 1 -3 LDH clay applications have been reported to range from catalysis, 4 -6 environmental, 7,8 biomedical, 9,10 polymer additives, 11 -13 among others.
LDH clays are quite versatile when compared to montmorillonite (Mnt) clays. Costa et al.
13
pointed out the following advantages that LDH clays have over the Mnt clays. LDH are reactive materials with inherent flame-retardation properties. Flexibility in organic modification, not limited to organic amines. The material can be customized to specific applications with high accuracy (e.g. homogeneous structure, chemical purity and chemical composition).
The single octahedral structure offers more reinforcing efficiency at low filler loading (i.e. more clay platelets per gram of clay when compared to Mnt).
In this report, we focus on LDH in polymer additive application, particularly in the polyamide 6 (PA6) matrix. A limited number of research groups that have published on PA6/LDH composites recently are summarised in Table 1.
Summary of the published work on PA6/LDH composites.

DBS: 4-dodecylbenzenesulfonate; PA6: polyamide 6; LDH: layered double hydroxide.
Due to the tuneable nature of LDH clays, conducting a direct comparison of the results from the different research groups would be a futile exercise. In addition, different preparation methods were used to produce the PA6/LDH composites. For example, Zammarano et al. 14 concluded that the LDH clays did not have significant nucleating effect, in terms of crystallite phase and rate of crystallisation on the PA6 matrix, while other groups reported a positive relationship between the presence of LDH clay and increased crystallisation rate of the PA6 matrix. 15,18,20 Improvement in the mechanical properties, 17,18 flame-retardation properties 15 and in situ polymerisation of ϵ-caprolactam to produce PA6/LDH nanocomposites 16 has been reported. In the case of Tabuani and co-workers, 18 they compared unmodified and organically modified LDH clay with other nanofillers and concluded that although organically modified LDH out-performed other nanofillers by improving the elongation at break of the PA6 matrix, Mnt clay and sepiolite gave better overall property improvement of PA6 matrix.
This study aims to contribute to the literature of PA6/LDH composites. In this vain, the thermal properties and rheological properties of the melt compounded PA6/LDH composites produced using organically modified and unmodified LDH clays at different levels were investigated.
Experiment
Materials
PA6 (Durathene B 30S, LANXESS) used in this work was supplied by Plastichem (Pty) Ltd, South Africa. PA6 has a density of 1.14 g cm−3 and melt volume flow rate of 110 cm3/10 min. Commercial LDH clays used in this work, and Perkalite™LD (unmodified, OH− interlayer anions) and Perkalite™FR100 (C16−18COO− fatty acid interlayer anions) from AkzoNobel were supplied by Sun Ace South Africa. PerkaliteLD (U-LDH) contains more than 90% of the Mg-Al hydroxide, density of 2.275 g cm−3, and PerkaliteFR100 (M-LDH) contains between 40% and 60% of Mg-Al hydroxide and between 40% and 60% of hydrogenated fatty acid and a density of 1.378 g cm−3. Alkanox 240, a processing antioxidant, was used in all mixtures at an average level of 0.27% (wt%) to stabilise the composite during melt compounding. All the materials were used as received, except for the polymer which was cryogenically pulverised using a Retzch™ 100 grinder, to a particle size of below 1 mm, before mixing with the nanofillers at different loadings; 1, 2.5, 5, 10, 15 and 25%.
Melt compounding
The mixtures of the pulverised PA6 and the LDH clays were dried in a vacuum oven at 50°C for 24 h, before melt compounding. The powders were tumble-mixed in the containers for 20 min before charging into the mixer head. A Thermo Haake PolyLab OS (USA) mixer, fitted with a twin rotor mixer head was used to compound the formulations. The mixer head was held at 240°C, rotor speed of 60 r min−1 and cycles of 8 min per mix, were used. The composite extrudates were then chopped into chips of less than 5 mm length. The chips were then pressed using Carver hydraulic press (model no. 3912) at 250°C for 10 min with a pressure of 2 tonnes to produce shapes and films for subsequent testing.
Transmission electron microscopy
The LDHs were analysed using transmission electron microscopy (TEM) to determine their chemical compositions and microstructure. The composites were analysed using TEM as well to determine their microstructure. For TEM, the LDH powders were ultrasonicated in absolute ethanol for 15 s and dispersed on carbon-coated copper (Cu) grids following a brief immersion in this medium. The PA6/LDH samples were sectioned under cryogenic conditions to prevent mechanical deformation which would have occurred during preparation at room temperature (RT). Solid polymer samples were mounted on aluminium pins and sectioned to a thickness of 70–90 nm at −120°C using a diamond knife of a Leica™ cryomicrotome. Sections were mounted on Cu grids prior to TEM observation.
All samples were viewed using a JEOL 2100 high-resolution TEM (Japan) fitted with an LaB6 filament at 200 kV. Calibrated images were capture using a Gatan Ultrascan (USA) digital camera. The elemental analysis of LDHs was done using the energy dispersive X-ray spectroscopy (EDS) detector of the instrument.
Scanning electron microscope
The morphology of LDH powders was determined by placing the powders on a conductive carbon tape and the samples scanned on the Auriga scanning electron microscope (SEM) (Germany). The moulded PA6/LDH samples were fractured cryogenically in liquid nitrogen to prevent mechanical deformation which would have occurred during preparation at RT. They were allowed to warm up before mounting on aluminium stubs using double-sided carbon tape and rendered conductive by sputter-coating with gold dust. Samples were viewed using a Carl Zeiss Auriga SEM (Germany) at an acceleration voltage of 1.3 kV for LDHs and 3 kV for PA6/LDH composites.
X-Ray diffraction
The powders of the nanofillers and 20-mm diameter × 1-mm thickness discs of the composites were analysed on the PANalytical™, X’Pert PRO X-ray diffraction (XRD) system (UK) using Cu K α1 radiation, at 45 kV and 40 mA. A scanning rate of 2° min−1 and scanning angle range of between 1° and 70° were used. XRD data of the composites were further analysed using deconvoluting software, PeakFit v4.12, to separate the peaks. The 2θ values of the α-phase crystals were estimated at (20.3 ± 0.2)° (α 1) and (23.6 ± 0.2)° (α 2); γ and the amorphous phase values were estimated at (21.4 ± 0.2)° and (22.5 ± 0.4)°, respectively.
Differential scanning calorimetry
Melting and crystallisation phenomena of the samples were analysed on the TA Instruments Q2000 differential scanning calorimetry (USA). For the dynamic experiments, the samples were heated from ambient to 270°C at 10°C min−1, held at 270°C for 5 min to remove their thermal histories. The samples were then cooled at 10°C min−1 to 50°C and subsequently heated to 270°C at 10°C min−1 for the second heating cycle. The melting data of the second heating cycles were used. The experiments were carried out under nitrogen atmosphere. The isothermal crystallisation half-time (t 1/2) experiments were also conducted by heating the samples from ambient to 270°C, held at this temperature for 5 min, thereby removing the thermal histories. The samples were then fast cooled to 200°C and held at this temperature for 30 min. The crystallisation time was measured from the point when the sample temperature reaches 200°C.
Thermogravimetric analysis
The thermal stabilities and ash residues of the clays and composites were determined using TA Instruments Q500 thermogravimetric analyser (USA). The samples, with an average weight of 5 mg, were heated from RT to 900°C at 10°C min−1 under nitrogen atmosphere.
Dynamic mechanical analysis
Dynamic mechanical properties of the composites were determined using TA Instruments Q800 dynamic mechanical analyser (USA). The samples (with average dimension: 18.5 mm (length) × 1.65 mm (thickness) × 9.8 mm (width)) were scanned from −60°C to 200°C at 3°C min−1 at 0.01% strain (c.a. 10-μm amplitude) and frequency of 1 Hz. The visco-elastic region of the composites was determined using a strain sweep method prior to sample analysis.
Rheological properties
Rheological (flow) properties of the composites were determined using TA Instruments ARES LS-2 rheometer (USA) with a parallel plate geometry. Samples of 1-mm thickness and 25-mm diameter were analysed in a frequency sweep mode (0.1–500 rad s−1) at 250°C and 10% strain, under constant nitrogen flow. The visco-elastic region of the composites was determined using a strain sweep method prior to sample analysis. The samples were analysed using oscillatory shear mode to determine their complex viscosity (η*), tan δ, storage modulus (G′) and loss modulus (G″).
Results and discussion
Transmission electron microscopy
TEM energy dispersive X-ray detector system (EDS) results are summarised in Table 2. The carbon, especially in the U-LDH, is from the counter balancing carbonate (
Chemical composition and the relative amount of the constituents of the fillers (wt%) as estimated by TEM EDS.

TEM: transmission electron microscopy; LDH: layered double hydroxide.
The LDHs were characterised by platelet particles with irregular shape. Lateral diameters of the LDH ranged from 0.7 μm to 4.6 μm with their thicknesses in the nanometer range.
The LDHs in the PA6 matrix displayed different morphologies. M-LDH displayed some increased spacing between the LDH interlayer spaces, pointing to some level of intercalation while U-LDH displayed tightly packed layers. These results suggested that intercalated PA6/M-LDH and immiscible PA6/U-LDH (microcomposite morphology) composites were produced.
Care should be taken with the interpretation of the TEM results and generalising them for the sample. The technique has a trade-off where only a small part of the specimen can be analysed at one time which may result in unrepresentative sampling hence the need to interpret the TEM results in combination with other techniques to confirm the morphology of the composites. 23 However, some evidence of even distribution of LDH particles in the PA6/U-LDH composite containing 2.5% clay is presented in Figure 1.

Transmission electron microscopic image of PA6/U-LDH composite containing 2.5% of U-LDH, showing evenly distributed LDH particles.
Scanning electron microscope
The morphologies of the LDHs used displayed no significant differences on their textures. This was not expected as Costa et al. 13 and Xu and Braterman 24 showed that organic modification altered the morphology, crystallite shape and texture of LDH clays. However, they conceded that this effect seems to be highly influenced by the surfactant being intercalated into the LDH.
The SEM micrographs of cryogenically fractured surfaces at 1000× magnification of the samples are shown in Figure 2. Figure 2(a) shows the morphology of the fractured surface of a neat PA6 while Figure 2(b) and (c) shows the fractured surfaces of PA6/M-LDH composites at 1 and 2.5% clay loading, respectively. Figure 2(d) to (f) shows the fractured surfaces of PA6/U-LDH containing 2.5, 5 and 10% clay, respectively. The samples displayed clear differences in the texture of their surfaces which indicated that the type of clay and clay content greatly influence the morphology of the matrix. The organic modification of the clay or lack thereof and clay content has been shown to influence the surface morphology of PA6 composites. 18 As expected, since the composites were cryogenically fractured, no shear bands or fibrous features were observed on the surfaces of the samples. It should be noted that no gas bubbles (porous morphology structure) were observed especially for the PA6/M-LDH (Figure 2(b) and (c), and Figure 3(a) and (c)) as the fatty acid has a lower decomposition temperature than the sample preparation temperature of 250°C. At low magnification (1000×), clay distribution could not be verified.

SEM micrographs at ×1000 magnification of the samples: (a) PA6, (b) PA6/M-LDH 1%, (c) PA6/M-LDH 2.5%, (d) PA6/U-LDH 2.5%, (e) PA6/U-LDH 5% and (f) PA6/U-LDH 10%.
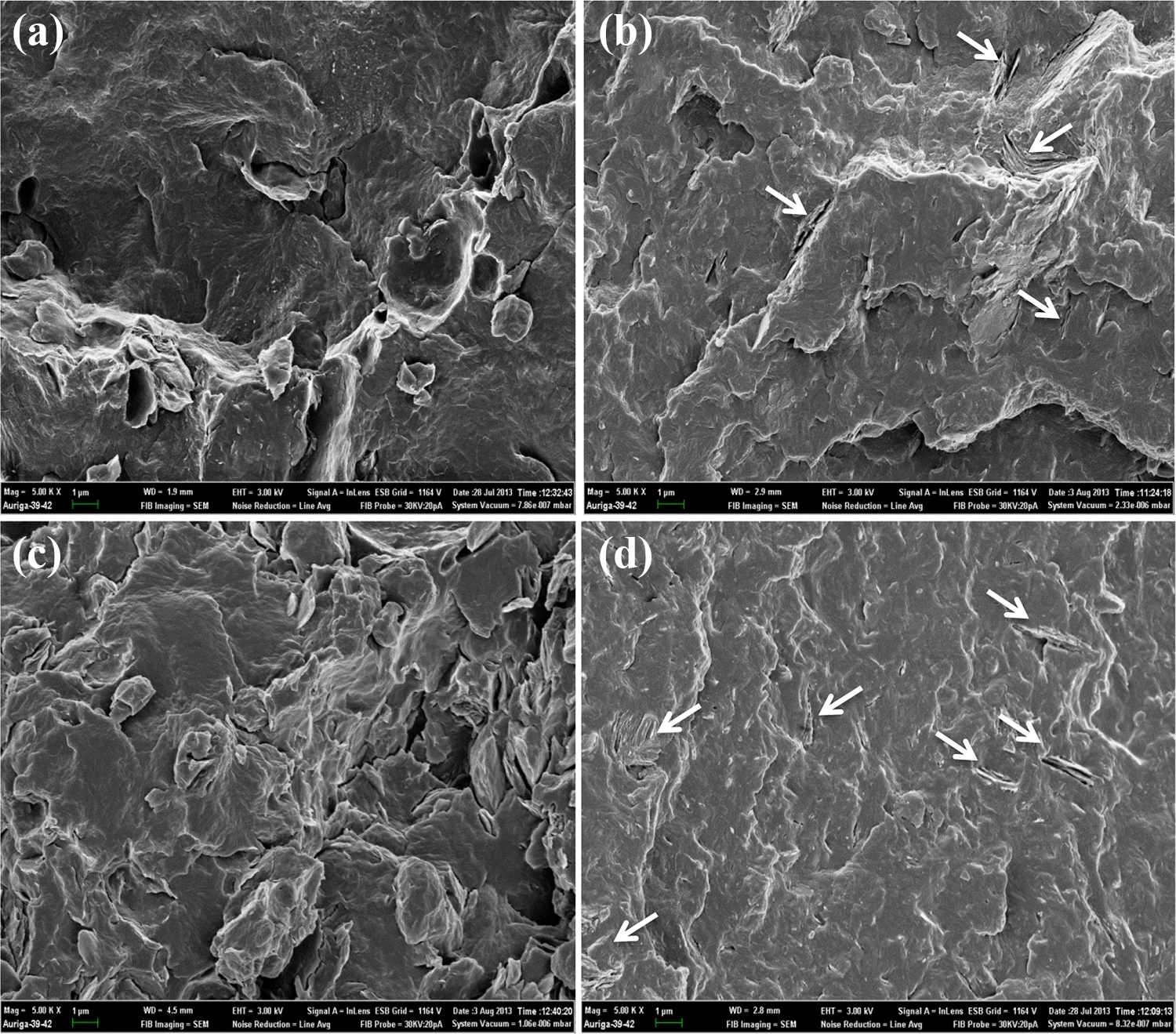
SEM micrographs at ×5000 magnification of the samples: (a) PA6/M-LDH 10%, (b) PA6/U-LDH 10%, (c) PA6/M-LDH 15% and (d) PA6/U-LDH 15%.
Figure 3(a) and (c) shows the micrographs of PA6/M-LDH at 10 and 15% clay loading, respectively, while Figure 3(b) and (d) shows the micrographs of PA6/U-LDH at 10 and 15% clay loading, respectively (5000× magnification). The clay particles in the PA6/M-LDH composites were hardly visible on the fractured surfaces of the samples, even at higher magnification. This suggests that the M-LDH clay was dispersed as stacks of few lamellae with thickness in nanometre scale, complete exfoliation could not be ruled out and the clay affinity of the matrix could be assumed. 18 On the other hand, evenly distributed clay sheets and tactoids are visible in the PA6/U-LDH composites, as shown by white arrows in Figure 3(b) and (d).
No evidence of small voids, which can be associated with clay-matrix de-bonding, were observed in the PA6/U-LDH samples, as the clay particles were still embedded in the PA6 matrix. This suggested an intimate surface interaction between the U-LDH clay and polymer matrix.
X-Ray diffraction
XRD patterns of the LDH are presented in Figure 4. U-LDH sample exhibited the characteristic 2θ reflections of Mg-Al are at 11.6° (003) and 23.4° (006), as reported by Costa et al. 13 and Yan-wu and Jun-qing. 16 A reflection at 18.3° (001) indicating the presence of Mg(OH)2 impurity can be seen on the U-LDH sample. 25 M-LDH displayed two strong reflections at lower diffraction angles (9.4° and 6.1°), indicating an increase in interlayer d-spacing due to the intercalated organic anions; in this case, its hydrogenated fatty acid, at different levels of intercalation. The 2θ = 11.6° reflection is still visible in the M-LDH (although weak), showing the presence of very low levels of unmodified LDH in the sample. The broad 2θ reflection between 18° and 25° is believed to be that of the organic component of the nanofiller.

X-Ray patterns of the modified and unmodified LDH.
PA6 2θ reflections for α 1 and α 2 crystals are displayed at 20.3° and 23.6°, respectively (Figure 5). Two observations were made on the PA6/M-LDH composites (Figure 5(a)). Firstly, the crystal reflections of the nanofiller were not present in the composites, indicating that the interlayer spaces of the LDH were completely separated as indicated by the disappearance of the LDH reflections (at 2θ = 9.4° and 6.1°) in the samples, an indication of complete exfoliation. However, at 15 and 25% M-LDH loadings, reflections at 2θ = 4.8° were present. This indicates that at these levels, the nanofiller could not be completely exfoliated as some ordered crystalline LDH structures were present in the matrix, possibly due to saturation.
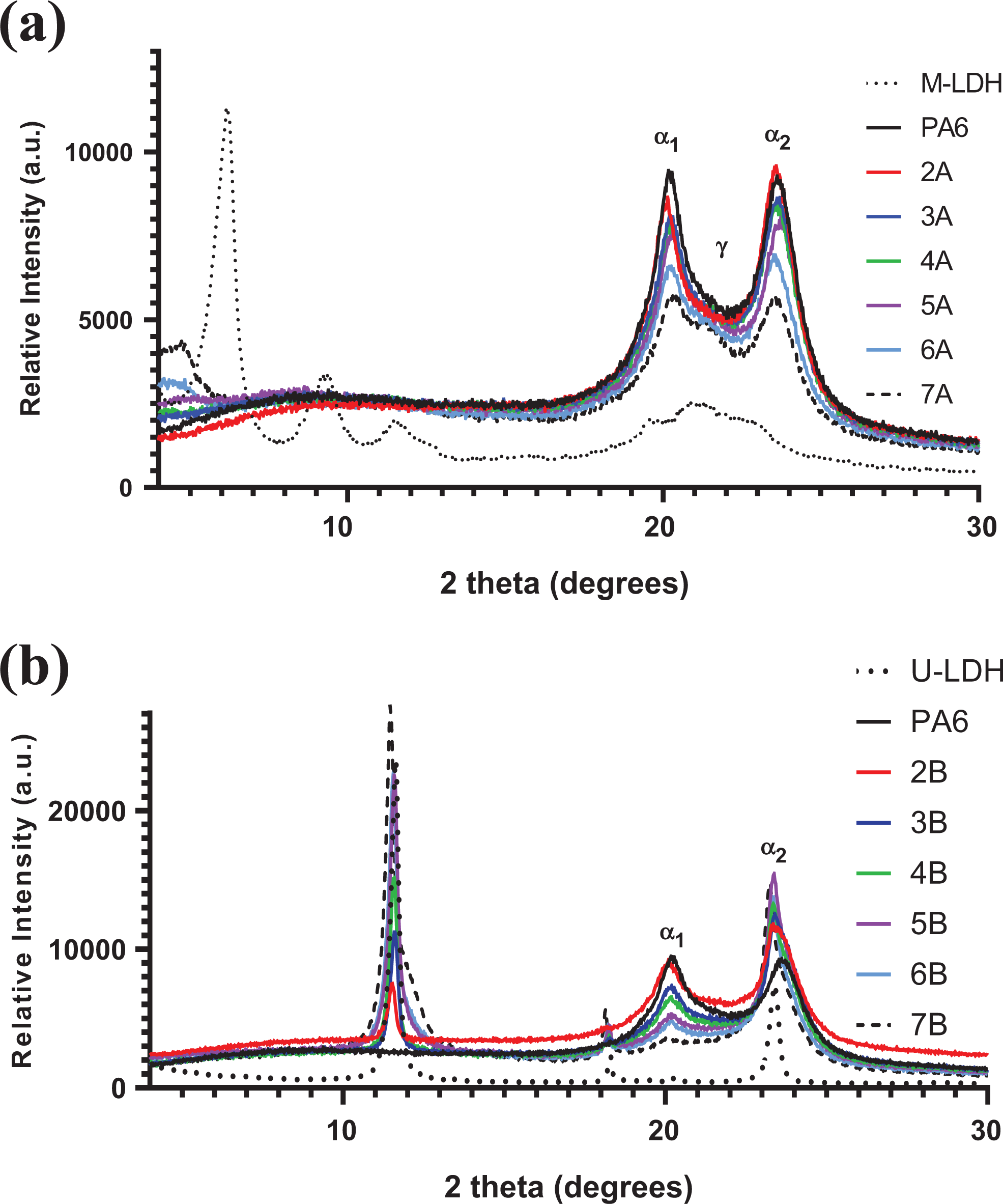
XRD patterns of the LDH and their respective composites: (a) PA6/M-LDH and (b) PA6/U-LDH.
Secondly, at LDH loading above 5%, the γ-crystallite peak of PA6 was observed at 2θ = 21.4°. 26 This was contrary to the results reported by Tabuani and co-workers, 18 and no γ-phase was detected in their PA6/M-LDH sample containing 5% clay. This indicated that several factors are responsible for the formation of γ-crystallites in PA6/LDH composites; however, a common one is the layered nanofiller exfoliation which results in polymer crystallising in between the nanofiller layers.
PA6/U-LDH composites displayed the following (Figure 5(b)): Firstly, the LDH characteristic peak (at 2θ = 11.6°) is visible in all samples and did not shift to smaller angles. This indicated that the polymer matrix did not affect the interlayer d-spacing of the LDH layers, that is, the polymer chains were not intercalated into the LDH interlayer spaces.
The 2θ = 11.6° peak broadened with increasing LDH content. Peak broadening and flattening of LDH in literature has been attributed to the reduction in crystallinity of the LDH due to either disintegration or dehydration induced by shear. 13,27 However, the sharpness of the LDH peak indicated that the broadening was influenced by other factors in the composites. It was proposed that the broadening of the 003 LDH peak could be attributed to polymer, possibly low-molecular-weight species or polymer chain ends, mildly intercalated into the LDH interlayer spaces. Secondly, the increase in LDH content decreased the intensity of the α 1 peak of PA6 (at 2θ = 20.3°), while the α 2 peak was masked by the 2θ = 23.4° peak of the LDH. This suggests that the increase in LDH content resulted in a decrease in the crystallisable component of the composites. No γ-peaks were observed in the PA6/U-LDH composites.
Differential scanning calorimetry
The addition of LDH to PA6 increased the onset temperature of crystallisation and the crystallisation peak temperature. The composites prepared for this work displayed narrower crystallisation peaks when compared to neat PA6 even at high LDH loadings, which indicated that the incorporation of LDH facilitated the matrix to crystallise in a manner which resulted in narrower crystallite dimension distributions. U-LDH displayed better heterogeneous nucleating effect on the PA6 matrix when compared to the M-LDH, even at high LDH content. This behaviour was attributed to the polymer not being intercalated in the interlayer spaces of the nanofiller, thus unrestricting the mobility of the polymer chains.
This phenomenon was evident in the PA6/M-LDH composites, where the increase in filler content reduced the onset of crystallisation temperature. At levels above 10% of M-LDH in PA6, it had a negative effect of lowering the onset temperature of crystallisation when compared to the neat PA6, this was also observed in the isothermal experiments.
The isothermal crystallisation results are in agreement with the dynamic experiments, that is, the U-LDH has a better heterogeneous nucleating effect on the PA6 composites when compared to the M-LDH, even at higher loadings of the nanofiller (Figure 6(a) and (b)). PA6/U-LDH composites displayed lower crystallisation half-life (t 1/2) when compared to the neat PA6. The t1/2, defined as the time it takes for the polymer spherulite to grow to 50% of its spherulites size during crystallisation. 28 M-LDH exhibited a negative influence on the crystallisation rate of the composites at 10% loadings and above. The differences in the nucleating effect of the two investigated LDHs is believed to be mainly controlled by the morphology, particle size and the distribution of nanofillers in the polymer matrix.

DSC isothermal crystallisation thermograms of the composites: (a) PA6/M-LDH and (b) PA6/U-LDH.
Thermogravimetric analysis
The thermal stability of unmodified LDH clay has been reported to be strongly influenced by the aluminium content 29 and intercalated inorganic anions 30 while that of organically modified LDH is influenced by the nature of the intercalated surfactant. 13 The presence of LDH clays in the PA6 matrix has been shown to negatively affect the thermal stability of the composites. This has been attributed to the basic nature of LDH clays, the release of interlayer water and dehydroxylation (in some cases in conjunction with surfactants), while the clay catalyses the nucleophilic attack of PA6. 13,14,17 Recently, Li et al. 20 suggested that LDH clay could be catalysing the degradation of PA6/LDH composite via a redox reaction mechanism. The composites prepared for this study displayed differences in their decomposition profiles (Figures 7 and 8), and the results are summarised in Table 3.

TGA thermograms of PA6/M-LDH composites: (a) weight loss curves and (b) DTG curves.

TGA thermograms of PA6/U-LDH composites: (a) weight loss curves and (b) DTG curves.
Summary of weight loss results at two specific regions and the ash residue of PA6/LDH composites.

PA6: polyamide 6; LDH: layered double hydroxide.
The authors proposed that low clay content would be associated with less water release and dehydroxylation products at elevated temperatures, and this would result in less initiation sites for the clay to catalyse the nucleophilic attack of the matrix. However, for the samples under investigation, no evidence of exfoliation was observed for PA6/U-LDH composites, and this suggests that the interaction between the clay and PA6 matrix (possibly hydrogen bonding on the clay surface and polymer chains) could be a plausible explanation for the improved thermal stability observed for these composites. On the contrary, for PA6/M-LDH composite, the increase in the surfactant concentration proved to have a negative impact on the thermal stability of the PA6 matrix. 14,15
Dynamic mechanical analysis
The viscoelastic properties of the composites, in the solid state, were determined using the dynamic mechanical analysis technique. The polymorphism of PA6 has been shown to have an effect on the mechanical properties of the polymer. In order words, the α- and γ-crystallites of PA6 were shown to have different stiffness (storage modulus) and damping (loss modulus) properties. 31 However, this effect is not always observed, since under normal processing methods, PA6 exhibits both crystallite forms. M-LDH clay displayed lesser reinforcing effect on the PA6 matrix when compared to U-LDH clay, especially in the temperature region between ambient and 200°C. At sub-ambient temperatures, PA6/M-LDH composites containing 5 and 10% clay had a negative effect on the stiffness of PA6. Interestingly, the 2% loading of M-LDH clay displayed similar stiffness to the PA6/M-LDH composite containing 25% clay in the temperature region between 90°C and 200°C, with an average increase of 18% in storage modulus when compared to neat PA6.
The poor reinforcing effect of M-LDH clay in the PA6 matrix was not expected, especially since the SEM experiments suggested that the clay was exfoliated in the polymer matrix. However, the results demonstrated that although the surfactant facilitated the distribution of clay in the matrix, it had a negative effect on the clay–matrix interaction. It was postulated that this was due to the incompatibility between the polymer and the surfactant for PA6/M-LDH composites, which resulted in poor interaction of the matrix and the clay, hence relatively little stress transfer occurred when the polymer was deformed. 32
On the other hand, for the PA6/U-LDH composites, only samples containing 1 and 5% U-LDH clay displayed similar stiffness as the pristine PA6. A ‘nano effect’ was observed in the sample containing 2.5% U-LDH clay, where the stiffness of the composite was improved at all test temperature range. It should be noted that at this clay level, in the temperature range between 90°C and 200°C, the stiffness increased by an average of 33% when compared to neat PA6. That is, above the glass transition temperature (T g) of the matrix, which is the α-transition, the modulus of the composites increased from 26% to 47% at 200°C, close to the matrix melting temperature (T m). This can be attributed to the difference in the mechanical properties of the polymer and the clay which remains rigid after the polymer has softened, post the T g as observed by Fornes Paul. 33
The composites displayed significant differences in their tan δ curves, with their shapes influenced by the dominant PA6 crystallite form (Figure 9(a) and (b)). Pristine PA6 displayed two peak maxima, at approximately T α1 39°C and T α2 71°C, corresponding to the α- and γ-crystallites of the polymer, respectively. Rotter and Ishida 34 pointed out that the anomaly in terms of the α-crystals (as a thermodynamically more stable form) having a lower T g can be attributed to the hydrogen bonding in the amorphous phase of the crystallites and not the crystallite integrity.
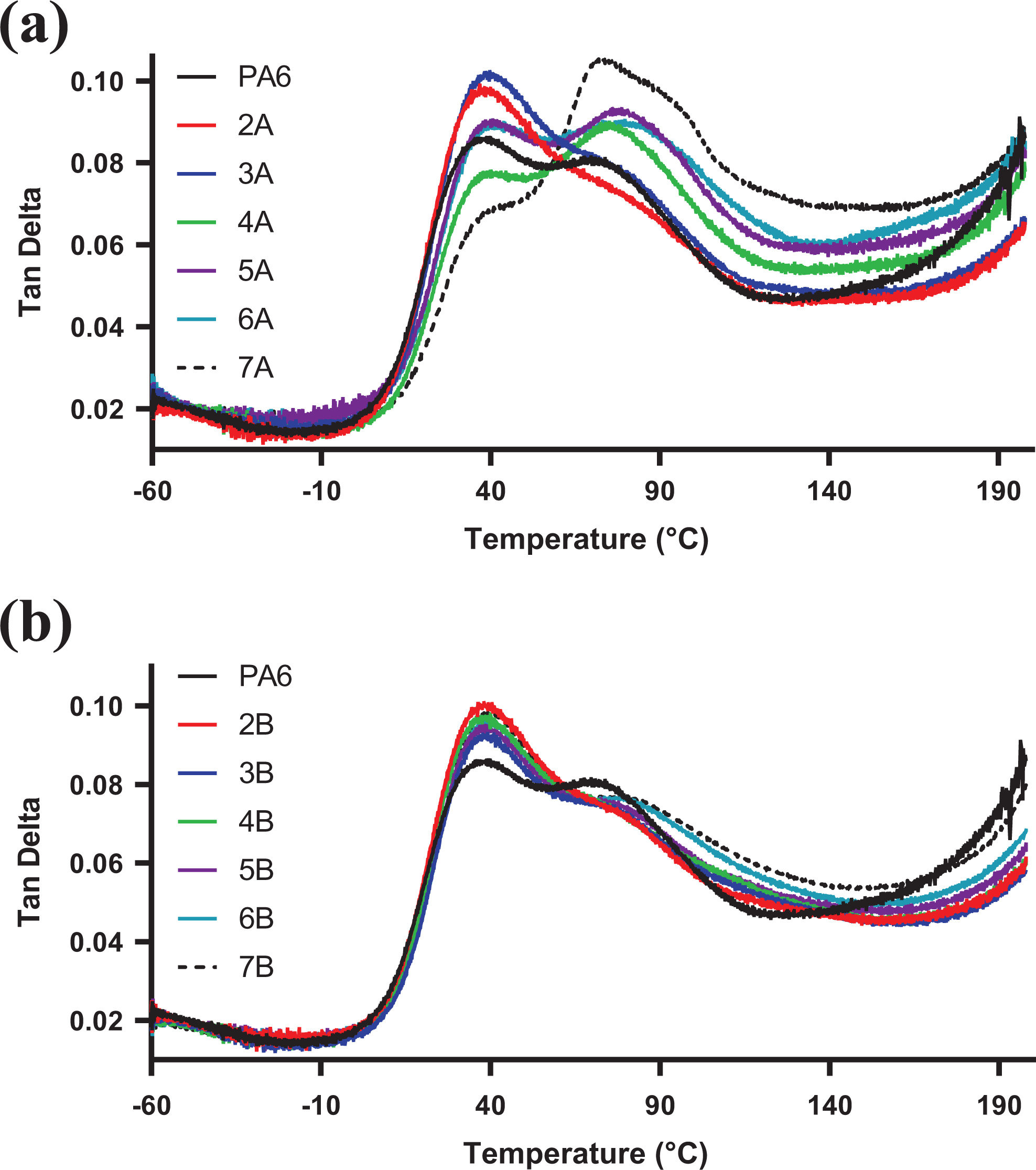
Tan δ curves of (a) PA6/M-LDH and (b) PA6/U-LDH composites.
PA6/M-LDH composites displayed an increase in the intensity of the T α2 peak, corresponding to the γ-crystallites, while a decrease in the intensity of T α1 peak corresponding to the α-crystallites, with increasing clay content. This was in agreement with the XRD results where the γ-crystallite diffraction peak was observed as the M-LDH clay content increased in the PA6 matrix. This confirms the fact that bulk of the polymer matrix crystallises between the clay galleries since the γ-form, which has been proposed to be responsible for improved mechanical properties of PA6/clay nanocomposites, dominates in the PA6/M-LDH samples. However, due to the lack of intimate interaction between the matrix and the nanofiller, the expected improvement in mechanical properties was not observed for these composites.
The PA6/U-LDH samples displayed a higher intensity of the T α1 peak, while the T α2 peak remained just a shoulder with increasing clay content when compared to the neat PA6. This confirmed the absence of γ-crystallite reflections on the XRD results of PA6/U-LDH composites.
The overall T g values of the composites did not differ significantly from that of the pristine PA6. The peak areas under the loss modulus curves of PA6/M-LDH composites did not show any particular trend with increasing clay content, while those of PA6/U-LDH increased with increasing clay content when compared to the neat PA6. However, an anomaly was observed for the composites of the two clays at 5% loading, where samples for both sets displayed areas that were less than that of the pristine PA6. The effect was more pronounced in the PA6/M-LDH sample. The PA6/U-LDH composites possessed better overall shock absorption capability when compared to PA6/M-LDH composites.
Rheology
The extent of clay–matrix interaction and the mesoscopic structure of the matrix primarily determine the rheological properties of PNCs. Size, shape, surface treatment, distribution and concentration of clay have been reported to have a significant effect on the rheological properties of PNCs. 35,36 The incorporation of LDH clays in the PA6 matrix increased the complex viscosity (η*) of the composites when compared to neat PA6, especially at low frequencies (ω; Figure 10(a) and (b)). Several factors, such as a strong clay–matrix interaction, exfoliation and dispersion of clay and the formation of network superstructures of clay layers in the melt, have been proposed to contribute to this behaviour of nanocomposites. These factors have been shown to be strongly influenced by the method of preparation (in situ polymerisation vs. melt-intercalation) of the samples. 37,38 PA6/M-LDH composites displayed significant increase in η* with increasing clay content. A Newtonian behaviour, similar to the neat PA6, was observed at lower frequencies for samples containing clay below 5%. Shear thinning behaviour of the PA6/M-LDH composites was more pronounced at clay loading above 10%.

Complex viscosity curves of (a) PA6/M-LDH and (b) PA6/U-LDH composites.
On the other hand, while the PA6/U-LDH composites displayed an increase in η* with increasing clay content, the slopes of their η* versus ω curves were constant over the investigated clay content range. Although evidence of shear thinning was observed, however, the frequency dependency was less pronounced when compared to that of PA6/M-LDH composites, especially at high clay loadings. A point of note on the PA6/U-LDH composites containing 1 and 2.5% clay, is that their η* displayed a yield behaviour at very low frequencies. This behaviour can be attributed to the breakage of network structures due to well dispersed clay (either exfoliated or intercalated), in order to align the clay layers in the direction of flow. 39
The results of storage modulus (elasticity–G′) and loss modulus (viscous–G″) of the composites indicated that both sets of composites had a solid-like behaviour, as shown by the monotonically increased values of G′ and G″ with increasing clay content. The effect is more pronounced in G′ curves than in G″ at low frequencies; this behaviour has been attributed to clay–matrix interaction and clay dispersion. 37 PA6/M-LDH displayed higher increase in the G′ and G″ values when compared to PA/U-LDH, indicating better M-LDH clay distribution than U-LDH. However, for the PA6/U-LDH composites, at 1 and 2.5% clay loadings, the results indicated a better clay dispersion and clay–matrix interaction when compared to the samples containing clay above 5% levels.
The ‘modified Cole–Cole’ plot, which is the logarithmic plot of G′ against log G″, has been used to evaluate the structural changes in the polymer matrix as a result of blending or filler/additive loading. In the modified Cole–Cole plot, when the additive (discontinuous phase) does not have an influence on the structure of the matrix at a given temperature, the resulting plots of the composite superimpose on the plot of the matrix. 38 Figure 11(a) and (b) shows the modified Cole–Cole plots of the PA6/M-LDH and PA6/U-LDH composites, respectively.

Modified Cole–Cole plots of (a) PA6/M-LDH and (b) PA6/U-LDH composites.
The incorporation of M-LDH into the PA6 matrix, significantly changed the microstructure of the composites’ melt, as shown by the curves of the composites which deviate significantly from that of PA6. Evidence of deviation was observed for PA6/U-LDH composites, although to a much lesser extent when compared to PA6/M-LDH composites. This indicated a lesser interaction of U-LDH and PA6 matrix in the melt state.
Conclusions
The clays used in the study had pronounced influence on the morphology, thermal, mechanical and rheological properties of the PA6 matrix. U-LDH clay generally imparted positive improvement in the PA6 matrix when compared to M-LDH. The organic modification of the clay proved to have a negative influence on the heterogeneous nucleation and thermal stability of PA6/LDH composites prepared. PA6/U-LDH had improved thermal stability, especially at clay levels below 5% and no deterioration was observed at higher clay loading. On the contrary, PA6/M-LDH composites displayed a negative trend, the thermal stability decreasing with increasing clay content. PA6/U–LDH composites also displayed superior stiffness and damping properties when compared to PA6/M-LDH composites. The clays promoted different crystallite forms in the PA6 matrix; PA6/U-LDH had predominantly α-crystallites while PA6/M-LDH had γ-crystallites. As expected, the incorporation of the clays in PA6 increased the complex viscosity of the matrix, and this was more pronounced for PA6/M-LDH composites. The M-LDH significantly altered the melt microstructure of PA6 as a result, PA6/U-LDH displayed better flow properties when compared to PA6/M-LDH composites.
Footnotes
Acknowledgements
The authors would like to thank the National Centre for Nano Structured Materials (CSIR) in particular Dr Thomas Malwela for SEM analysis and Dr James Wesley-Smith for TEM analysis, Applied Chemistry Department (University of Johannesburg), Sun Ace South Africa, Plastichem (Pty) Ltd, Sarah Mokwena for rheology analysis and last but not the least, the Customer Support Team (Sasol Polymers).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This work was supported by the Polymer Division (Tshwane University of Technology), National Research Foundation (NRF) under grant no. 86730.