
Abstract
Mesenchymal stem cells (MSCs) are recognized for their capacity to modulate immune responses, including those directed against tumors. In this study, we investigated the temporal effects of MSCs administration on anti-tumor immunity in a murine 4T1 breast cancer model. BALB/c mice were intraperitoneally injected with MSCs either 24 h (MSC1d) or 14 days (MSC14d) after orthotopic implantation of 4T1 mammary carcinoma cells. Early MSC administration (MSC1d) exhibited changes in immune cell phenotypes consistent with enhanced antitumor potential, including increased activity of natural killer (NK) cells, dendritic cells (DCs), macrophages, and T lymphocytes. These immunological changes correlated with reduced tumor growth and prolonged survival. Mice in the MSC1d group exhibited elevated serum levels of pro-inflammatory and anti-tumor cytokines (TNF-α, IFN-γ, IL-6, and IL-17), alongside decreased concentrations of immunosuppressive cytokines (TGF-β and IL-10). Tumor tissue analysis revealed increased infiltration of NK cells expressing markers associated with antitumor activity (IFN-γ-producing CD178⁺), CD80⁺/CD86⁺/I-A⁺ TNF-α-producing DCs, Th1-type CD4⁺ T cells, and Granzyme B-expressing CD8⁺ cytotoxic T lymphocytes (CTLs). Additionally, spleens of MSC1d-treated mice displayed significantly elevated populations of CD11c⁺ DCs, TNF-α/IFN-γ-secreting NK cells, CD4⁺ Th1 and Th17 cells, and CD8⁺ CTLs expressing markers associated with cytotoxic function (TNF-α, IFN-γ, and IL-17). Conversely, late MSCs administration (MSC14d) was associated with immunosuppression. Tumors from MSC14d-treated mice showed a decreased presence of IFN-γ⁺ and IL-17⁺ NK1.1⁺ cells, F4/80⁺ macrophages, IL-12⁺ DCs, and cytotoxic T cells. Spleens from these mice revealed a significant expansion of regulatory T cell (Treg)-like populations, including CD25⁻, FoxP3⁻, CD25⁺FoxP3⁻ cells, and TGF-β/IL-10-producing CD3⁺ and CD4⁺ T cells. Furthermore, serum levels of immunosuppressive mediators TGF-β and vascular endothelial growth factor (VEGF) were significantly elevated in the MSC14d group. Collectively, these findings demonstrate that the immunomodulatory effects of MSCs on breast cancer are highly dependent on the timing of their administration. Mesenchymal stem cells delivered during early tumor development enhance phenotypes consistent with antitumor potential and suppress tumor progression, whereas administration during later stages promotes immune evasion and tumor growth.

Introduction
Breast cancer remains a significant global health burden, with approximately 2.3 million new cases diagnosed annually and a global mortality rate of 6.9% 1 . Despite notable advances in therapeutic strategies—including surgery, radiotherapy, chemotherapy, endocrine therapy, and immunotherapy—the prognosis for patients with metastatic breast cancer remains poor, with fewer than 30% achieving 5-year survival 2 . Persistent rates of incidence and mortality underscore the limitations of current treatments, particularly in light of tumor heterogeneity, which has been identified through omics-based research as a key contributor to therapeutic resistance and disease recurrence 2 . These findings highlight the urgent need for alternative and more personalized therapeutic approaches for patients unresponsive to conventional treatments. Mesenchymal stem cells have emerged as promising candidates for cell-based therapies due to their regenerative capacity, immunomodulatory properties, and anti-inflammatory potential 3 . These multipotent stromal cells, which are present in nearly all connective tissue-rich organs, display a fibroblast-like morphology and play a central role in tissue repair and immune regulation 4 . Mesenchymal stem cells are actively being investigated in preclinical and clinical trials for a range of conditions 3 . Notably, MSCs are integral components of the tumor stroma and are known to interact dynamically with other stromal elements, including lymphocytes, macrophages, myeloid-derived suppressor cells (MDSCs), vascular endothelial cells, and tumor-associated fibroblasts (TAFs), often promoting tumor progression 5 . A key characteristic of MSCs is their ability to migrate and home to sites of tissue damage and tumors, a process mediated by cytokine and chemokine gradients and facilitated by matrix metalloproteinases (MMPs), which degrade the basement membrane and support tissue infiltration 6 . Upon systemic administration, MSCs preferentially localize to the tumor microenvironment, where they modulate tumor growth and immune responses via direct cell-cell interactions (juxtacrine signaling) and through paracrine mechanisms, notably involving extracellular vesicles (EVs) 7 . Mesenchymal stem cells can act as cellular delivery vehicles for anticancer agents, including pro-inflammatory cytokines, microRNAs, and cytotoxic molecules, thereby contributing to tumor inhibition 8 . These cells also profoundly affect the phenotype and function of various immune cells- modifying antigen presentation by macrophages and dendritic cells (DCs), enhancing or suppressing the cytotoxicity of natural killer (NK) and CD8⁺ T cells, and altering cytokine production by CD4⁺ T helper cells 9 . Interestingly, MSCs exhibit dual and often contradictory roles in breast cancer. Some studies report MSCs-induced tumor promotion, characterized by increased proliferation, metastasis, epithelial-to-mesenchymal transition (EMT), and therapeutic resistance10,11. Conversely, other findings, including those by Zhou et al. 12 and Zhang 13 , suggest that MSCs possess anti-tumor properties, such as proliferation inhibition, apoptosis induction, and enhanced immune cell infiltration into tumors. These divergent outcomes may reflect the influence of tumor heterogeneity, inter-donor MSCs variability, and experimental parameters such as timing, dosage, and combination with other therapies 14 . A key clinical concern arises in the context of MSCs-based therapies administered to patients with undiagnosed, early-stage breast cancer for unrelated conditions (eg, osteoarthritis and diabetic ulcers)15,16, raising questions about the potential impact of MSCs on tumor progression. Similarly, in patients with advanced-stage breast cancer, the effects of MSCs on tumor dynamics remain unclear.
Given these uncertainties, the current investigation was conducted to assess whether the timing of MSCs administration critically influences anti-tumor immunity and tumor progression, using a murine 4T1 breast cancer model.
Materials and methods
Cell lines
In this study, the murine mammary carcinoma cell line 4T1 (American Type Culture Collection, ATCC, Manassas, VA, USA) was employed. This cell line was derived from a spontaneously occurring mammary tumor in a BALB/c mouse, serving as an established model for stage IV breast cancer research. In this study, we used commercially available BALB/c mouse bone marrow–derived mesenchymal stem cells obtained from Cell Biologics (Cat. No. 1308071612; Lot No. 5043), and were characterized by the supplier by immunofluorescence staining for CD44, Sca–1, and CD29. Both cell types were cultured in Dulbecco Modified Eagle Medium (DMEM) supplemented with
Animal model
This study utilized female BALB/c mice aged 8 to 10 weeks. Mice were accommodated in the animal facility of the Faculty of Medical Sciences, University of Kragujevac, Serbia. All experimental procedures were conducted in accordance with the Principles of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals. The Animal Ethical Review Board of the University of Kragujevac authorized the study protocol. Mice were maintained under a controlled environment with a 12-h light/dark cycle and had access to standard laboratory chow and water ad libitum. A minimum of nine mice were included in each experimental group. In this study, only female BALBc mice that were 8 to 10 weeks of maturity that developed tumors were included in the study. Animals excluded from the analysis were those from the experimental group that died prior to the day of sacrifice. The rationale is that the volume and weight of the mammary tumors were assessed solely for the mice that lived until the completion of the experiment. We used randomization to allocate experimental units to control and treatment. In order to reduce the pain and suffer of animals, mice were euthanized by cervical dislocation. We used the ARRIVE reporting guidelines to meet all the criteria for the study involving animals 17 .
Tumor induction and MSCs injection
4T1 cells (5 × 104) were suspended in 50 µl of phosphate-buffered saline (PBS) and orthotopically injected into the fourth mammary fat pad of BALB/c mice. One day postinjection, the mice were assigned to four experimental groups (n = 9 per group). The first experimental group (4T1+MSC1d) received intraperitoneal (IP) injections of MSCs (5 × 105 cells suspended in 200 µl of PBS) 1 day after 4T1 cell injection. The second group (4T1+MSC14d) received MSCs (5 × 105 cells in 200 µl PBS) 14 days following 4T1 injection. The third and fourth groups (4T1+PBS1d and 4T1+PBS14d) received 200 µl PBS IP at 1 and 14 days post-4T1 cell injection, respectively. All mice were euthanized 35 days post-4T1 cell injection.
Tumor development and progression monitoring
Tumor volume was monitored once tumors became palpable, measured every 3 days using electronic calipers. Tumor volume was calculated with the formula: V = 4/3π*a/2*b/2*c/2 (a = length, b = width, c = thickness) where a, b, and c are the tumor length, width, and thickness, respectively 18 . Metastatic spread was assessed by a blinded pathologist in hematoxylin and eosin (H&E)-stained paraffin-embedded tissues from the lung, liver, and brain. Pathohistological scores were assigned based on metastasis presence and extent in these organs, as well as isolated breast tumors.
Serum cytokine analysis
Blood samples were collected on days 1, 14, and 35 post-4T1 cell injection. Serum was stored at −20°C for subsequent analysis. Serum cytokine levels of TNF-α, IFN-γ, IL-6, IL-17, TGF-β, and IL-10 were quantified using enzyme-linked immunosorbent assay (ELISA) kits specific for murine cytokines (R&D Systems, Minneapolis, MN, USA) following the manufacturer’s protocols 19 .
Tumor-infiltrating leukocyte isolation
4T1 tumors were excised in their entirety, including surrounding skin. Tumors were weighed, measured, and photographed before being cut into small pieces and digested in 5 ml of DMEM containing 1 mg/ml collagenase I, 1 mM ethylenediaminetetraacetic acid (EDTA), and 2% FBS (Sigma-Aldrich, Munich, Germany). The tissue underwent incubation at 37°C for 2 h with trypsin and DNase I (Roche Diagnostics), subsequently being filtered through a 40-μm nylon filter. The resultant single-cell suspension underwent flow cytometry analysis 20 .
Spleen cell isolation
At 35 days post-4T1 cell injection, mice were euthanized, and spleens were harvested. Spleens were processed through a cell strainer (BD Pharmingen, USA) into a 50-ml tube containing 5-ml RPMI-1640 medium with 10% FBS. The cell suspension was centrifuged at 350 × g for 5 min, and the supernatant was discarded. The cell pellet was resuspended in 5 ml of erythrocyte lysis buffer (EDTA, NaHCO₃, NH₄Cl in ddH₂O), incubated on ice for 5 min, and then washed with RPMI-1640 + 10% FBS. To remove histiocytes, the cell suspension was filtered again. The final splenocyte suspension was prepared for flow cytometry 21 .
Flow cytometry and intracellular cytokine staining
Flow cytometry was performed to analyze cell surface and intracellular markers on splenocytes and tumor-infiltrating leukocytes. Cells (1 × 106) were stained with fluorochrome-conjugated monoclonal antibodies against CD3, CD4, CD8, CD11c, NK1.1, CD86, I-A, granzyme B, and Fas ligand (FasL) according to the manufacturer’s protocols. For intracellular cytokine staining, cells were stimulated with 50 ng/ml Phorbol 12-myristate 13-acetate (PMA) and 500 ng/ml ionomycin for 5 h, followed by GolgiStop addition. Cells were fixed, permeabilized, and stained with antibodies against TNF-α, IFN-γ, and IL-17. Flow cytometry was performed using a BD FACSCalibur system and analyzed with Flowing Software 22 .
Statistical analysis
Statistical analysis utilized SPSS version 21. The Kolmogorov-Smirnov test was employed to evaluate the normality of the data. The Student’s t-test was utilized to assess differences between groups. Data are expressed as mean ± standard error of the mean (SEM). Statistical significance was defined as P < 0.05. Tumor volumes were measured repeatedly in the same animals; therefore, these data are repeated-measures observations with within-subject correlation. While pairwise comparisons at selected time points were performed here, repeated-measures analysis of variance (ANOVA) or linear mixed-effects models that account for within-mouse correlation across time points will be used in future analyses.
Results
The growth and progression of breast cancer are reduced by the administration of MSCs 24 h after tumor induction
Initially, the effects of IP administration of MSCs on breast cancer progression were assessed. As demonstrated in Fig. 1a, tumors became palpable in both experimental groups 9 days following tumor induction. By day 21, the mean tumor volume in mice treated with MSCs 24 h after tumor induction (4T1+MSC1d) was markedly diminished in comparison to the control group treated with PBS (4T1+PBS1d) (P < 0.05; Fig. 1a), indicating that early MSCs administration effectively inhibited tumor growth. This inhibitory effect persisted through day 35, with both the average tumor volume and weight remaining significantly lower in the 4T1+MSC1d group relative to controls (Fig. 1b, c). Additionally, tumor measurements at day 35 confirmed this trend, with tumors in the MSC-treated group significantly smaller than those in the PBS-treated group (Fig. 1d).

The timing of MSCs administration affects the effect of MSCs on breast cancer progression. Different day post tumor-induction, tumor volume measurements revealed delayed tumor growth in mice treated with 4T1+MSC1d beginning on day 21 (a). At day 35, the average tumor volume (b, d) and tumor weight (c) were significantly lower in 4T1+MSC1d-treated mice than in 4T1+PBS1d-treated animals. Until the day 35 of the experiment, 4T1+MSC1d-treated mice had the higher survival rate compared to 4T1+PBS1d-treated mice (e). The survival difference between experimental groups was not statistically significant (“ns”). The weight loss of MSC-treated, 4T1-bearing mice decreased, as evidenced by the average animal weight at various days following tumor induction compared to 4T1+PBS group (f). Values shown as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
Survival analysis further revealed that the timing of MSCs administration played an important role in therapeutic efficacy. Mice receiving MSCs 24 h postinduction exhibited improved survival rates compared to those receiving only PBS, although the difference was not statistically significant (P > 0.05; Fig. 1e). By day 14, both groups showed an increase in body weight, with a slightly greater increase observed in the PBS group; however, this difference did not reach statistical significance (P > 0.05; Fig. 1f).
Liver damage was evaluated histopathologically, including assessment of hepatocyte degeneration, necrosis, and periportal and portal inflammation 23 (Fig. 2a, g). Mice in the 4T1+PBS group exhibited pronounced portal infiltration, inflammatory changes, and localized necrosis, whereas the 4T1+MSC1d group displayed notably milder pathology, including Kupffer cell hyperplasia and moderate hepatocyte degeneration. Normal hepatic architecture was observed in healthy controls. Quantitative histopathological scoring revealed significantly reduced liver damage in the MSCs-treated group compared to controls (P < 0.001; Fig. 2a). Lung tissue was examined for alveolar wall thickening, atelectasis, emphysema, alveolitis, fibrosis, pneumonitis, and pneumocyte hyperplasia or hypertrophy 24 . The 4T1+PBS1d group showed evidence of compensatory emphysema and widespread structural damage, while only one-third of the 4T1+MSC1d mice exhibited mild alveolar alterations. Lung histology was unremarkable in healthy controls. Overall lung damage was significantly lower in the MSCs-treated group compared to the PBS group (P < 0.001; Fig. 2b). Brain tissue was analyzed for edema, activation of glial cells, reactive gliosis, and necrosis 25 . No pathological changes or metastatic deposits were observed in the 4T1+MSC1d group or in healthy mice (Fig. 2g). Brain damage scores were notably decreased in the animals treated with MSCs in comparison to those in the 4T1+PBS1d group (P < 0.001; Fig. 2c), which exhibited the highest level of brain pathology among all groups (P < 0.001; Fig. 2c).

The timing of MSC administration affects the effect of MSCs on breast cancer progression. Liver, lung, brain, and tumor pathohistological scores (a–d). Lung and liver metastases (e–f). Light-microscopic pictures through liver, lungs, brain, and tumors (H&E staining; magnification ×40): circle denote metastases, solid arrows signify necrosis, and rectangles represent lymphocyte infiltrates) (g). Serum samples from mice treated with 4T1+MSC1d exhibited significantly elevated levels of pro-inflammatory and anti-tumor cytokines, including IFN-γ (h), TNF-α (i), IL-6 (j), and IL-17 (k). In contrast, these samples showed a notable reduction in immunosuppressive cytokines IL-10 (l) and TGF-β (m) when compared to serum samples from mice treated with 4T1+PBS1d. Values shown as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
Histological analysis of primary tumors indicated central, localized necrosis and modest perivascular lymphocytic infiltration in tumors 26 from the MSCs-treated group (Fig. 2d). In contrast, tumors in the PBS group were composed of malignant cells with polymorphic, hypertrophic nuclei, abundant mitoses, and absence of necrosis, reflecting a more aggressive phenotype. Quantitative evaluation confirmed that tumor damage was significantly reduced in the MSCs-treated group (P < 0.001; Fig. 2d). Metastatic burden was also assessed. The number of lung metastases was significantly lower in the 4T1+MSC1d group compared to the PBS-treated group (P < 0.05; Fig. 2e). Although liver metastases were more frequent in the PBS group, it must be noted that the difference did not reach statistical significance (P > 0.05; Fig. 2f). No brain metastases were detected in any of the experimental groups. Cytokine profiling revealed that the MSCs-treated group exhibited significantly elevated serum levels of pro-inflammatory and anti-tumor cytokines, including IFN-γ, TNF-α, IL-6, and IL-17 (P < 0.05 to P < 0.001; Fig. 2h–k). Conversely, levels of immunosuppressive cytokines, specifically IL-10 and TGF-β, were significantly decreased in the MSCs-treated group (P < 0.05 and P < 0.001, respectively; Fig. 2l, m). These results suggest that IP administration of MSCs during the early phase of tumor development enhances anti-tumor immune responses and mitigates tumor-associated tissue damage.
MSCs applied 24 h after 4T1 induction significantly enhance the antitumor and cytotoxic potential of NK, T cells and enhance the capacity of DCs to present tumor antigens, suppressing tumor progression and growth
Compared to tumors from 4T1+PBS1d-treated mice, those from 4T1+MSC1d-treated animals exhibited a significantly higher infiltration of innate immune cells critical for antitumor defense, specifically cytotoxic NK cells and DCs. Analysis of tumor-infiltrating lymphocytes revealed that administration of MSCs 24 h post-4T1 cell inoculation substantially augmented the number of NK1.1⁺ NK cells within the tumor microenvironment (P < 0.05; Fig. 3a). Furthermore, the population of Fas ligand (FasL)- expressing NK cells (P < 0.05; Fig. 3b) and those producing interferon-gamma (IFN-γ) (P < 0.05; Fig. 3c) was markedly elevated in the 4T1+MSC1d group, indicating that MSCs treatment enhanced the cytotoxic and antitumor functions of NK cells in tumor- bearing mice. In addition to NK cell activation, a significant increase in tumor-infiltrating CD11c⁺ DCs was observed in MSCs-treated animals (P < 0.05; Fig. 3d). These cells expressed higher levels of the co-stimulatory molecule CD86 (P < 0.05; Fig. 3e) and MHC class II molecules (I-A) (P < 0.05; Fig. 3f). Moreover, a greater number of CD11c⁺ DCs co-expressing I-A and producing TNF-α were detected in the MSCs-treated group (P < 0.05; Fig. 3g). These findings suggest that MSCs administration enhances the antigen-presenting capacity and proinflammatory profile of DCs, thereby promoting effective activation of T cell-mediated antitumor responses. Adaptive immune responses were also significantly improved in the MSCs-treated group. Specifically, there was a marked increase in the number of tumor-infiltrating CD4⁺ Th1 cells (P < 0.05; Fig. 3h), as well as IFN-γ-producing CD4⁺ Th1 cells (P < 0.05; Fig. 3i) in mice treated with MSCs. Furthermore, the number of CD8⁺ cytotoxic T lymphocytes (CTLs) was significantly elevated in the 4T1+MSC1d group compared to PBS-treated controls (P < 0.05; Fig. 3j). These CTLs also exhibited increased expression of cytotoxic effector molecules FasL and Granzyme B (P < 0.05; Fig. 3k, l), indicating that MSCs treatment potentiated the cytolytic activity of CD8⁺ T cells within the tumor microenvironment.

Being administered 24 h after 4T1 induction, MSCs dramatically improved antitumor immunity generated by NK, DCs, and T cells, and inhibited tumor growth and progression. In the tumors of 4T1+MSC1d-treated mice there was significantly greater number of NK1.1+NK cells (a), FasL- expressing and IFN-γ producing NK cells (b, c), CD11c+DCs (d), CD86- (e) and I-A-expressing (f) and I-A-expressing, TNF-α producing CD11c+DCs (g), CD4+Th1+ (h) and IFN-γ- producing CD4+Th1 cells (i), CD8+CTLs (j), FasL- (k) and Granzyme B-expressing CD8+CTLs (l) in comparison to 4T1+PBS1d-treated mice. Values shown as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
Administration of MSCs 24 h post-4T1 induction significantly altered the cellular composition of the spleen in mice, which increased DC antigen presenting ability and NK and T cell numbers suppressing tumor progression and growth
We also analyzed immune cells isolated from the spleen of mice and discovered that the spleens of 4T1+MSC1d-treated mice exhibited a significantly higher number of innate immune cells that are essential for antitumor immunity, specifically DCs and NK cells, in comparison to the spleens of 4T1+PBS1d-treated mice. The significantly higher number of CD11c+DCs (P < 0.001; Fig. 4a) and TNF-α producing CD11c+DCs (P < 0.001; Fig. 4b) in spleens of 4T1+MSC1d-treated mice compared to 4T1+PBS1d-treated mice, suggests that MSCs enhanced capacity of antigen-presenting cells for optimal activation of T cell-driven antitumor immune response. Notably, the cellular composition of spleens collected from 4T1+PBS1d and 4T1+MSC1d-treated mice demonstrated that MSCs injected 24h after 4T1 induction increased the total number of tumor-infiltrating cytotoxic TNF-α and IFN-γ producing (P < 0.001; Fig. 4c, d) NK cells in the spleens of 4T1+MSC1d-treated animals. The presence of NK cells in the spleens of mice treated with 4T1+MSC1d revealed MSCs administered 24 h post-4T1 induction enhanced the cytotoxic and antitumorigenic functions of NK cells in tumor-bearing mice. In the spleens of animals treated with MSCs 24 h after 4T1 induction, there was a significantly increased number of antitumorigenic IFN-γ and TNF-α producing CD3+ cells (P < 0.001; Fig. 4e, f), as well as TNF-α, IFN-γ producing CD4+Th1 cells (P < 0.05; Fig. 4g, h), and IL-17- producing CD4+Th17 cells (P < 0.001; Fig. 4i) compared to the group of 4T1+PBS1d-treated mice. Also, in the spleens of animals that received MSCs 24 h after 4T1 induction, it was noticed that the total number of CD8+CTLs (P < 0.05; Fig. 4j), as well as TNF-α, IFN-γ, and IL-17-producing CD8+CTLs (P < 0.05; Fig. 4k–m) was significantly higher compared to 4T1+PBS1d group of animals.

MSCs increased the number of DC, NK, and T cells in mice’s spleen 24 h after 4T1 induction, inhibiting tumor development and progression. In the spleens of 4T1+MSC1d-treated mice there was a significantly greater number of CD11c+DCs (a) and TNF-α producing CD11c+DCs (b), TNF-α and IFN-γ producing NK cells (c, d), IFN-γ and TNF-α producing CD3+ cells (e, f), TNF-α, IFN-γ producing CD4+Th1 (g, h), as well as IL-17-producing CD4+Th17 cells (i), CD8+CTLs (j), as well as TNF-α, IFN-γ, and IL-17-producing CD8+CTLs (k, l, m) in comparison to 4T1+PBS1d-treated mice. Values shown as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
The administration of MSCs 14 days after 4T1 tumor induction increases the growth and progression of breast cancer
Fig. 5a illustrates that tumors became visible in both groups 9 days posttumor induction. On day 23 (9 days post-MSC injection), the mean tumor volume of 4T1+MSC14d-treated animals exceeded that of 4T1+PBS14d-treated mice, although the difference was not statistically significant (P > 0.05; Fig. 5a). At day 35, the mean volume and mass of tumors excised from 4T1+PBS14d-treated mice were markedly inferior to those from 4T1+MSC14d-treated subjects (P < 0.001; Fig. 5b, d and P < 0.05; Fig. 5c), thereby substantiating that MSCs administered 14 days posttumor induction significantly expedited 4T1 tumor growth and advancement. The timing of MSC injection was crucial for its impact on the survival of 4T1-bearing mice. While the lowest survival rate was observed in 4T1+MSC14d treated mice, there was a trend in higher survival rate in the breast cancer-bearing animals that received PBS 14 days after tumor induction (P > 0.05; Fig. 5e). At day 26 until day 33, survival rate was lower in 4T1+MSC14d treated group, compared to 4T1+PBS 14d treated group, but this difference was not statistically significant (Fig. 5 (P > 0.05; Fig. 5e)). On the 14th day, an increase in average body weight was detected in both groups of animals, with the biggest rise occurring in the 4T1+MSC14d group, although this difference was not statistically significant (P > 0.05; Fig. 5f).

The timing of MSCs administration affects the effect of MSCs on breast cancer progression. On day 21, tumor volume measurements indicated that the proliferation of breast cancer in animals treated with 4T1+MSC14d was accelerated (a). At day 35, the average tumor volume (b) and tumor weight (c) were significantly larger in 4T1+MSC14d-treated mice compared to 4T1+PBS14d-treated mice (d). Until day 35 of the experiment, 4T1+MSC14d-treated animals had the lowest survival rate (e). The survival difference between groups was not statistically significant (“ns”). Average animal weight of mice from 4T1+MSC14d group show an increase in average weight at various days following tumor induction compared to 4T1+PBS14d group (f). Values shown as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
In the group of mice with 4T1+MSC14d, there was moderate portal space infiltration, hyperplasia of Kupffer cells, and parenchymal hepatocyte degeneration. In terms of pathohistological score, the highest pathohistological score of liver damage was observed in the 4T1+PBS animal group compared to 4T1+MSC14d (P < 0.001; Fig. 6a). In addition to inflammation, regions of localized necrosis were identified in the group of animals only with 4T1+ PBS14d (Fig. 6g). In the 4T1+MSC14d mouse group, areas of alveolitis and compensatory emphysema were observed, while in the 4T1+PBS14d mouse group, only compensatory emphysema was found. In the group of healthy mice, there was normal lung histomorphology (Fig. 6g). Furthermore, the greatest lung damage was observed in the 4T1+PBS14d mice group, compared to 4T1+MSC14d group (P < 0.001; Fig. 6b). We also analyzed pathohistological score of brains in this study (Fig. 6c). In the 4T1+ MSC14d group of mice, rare granular cells were found, whereas in the group of mice with mammary carcinoma alone, focal reactive gliosis of the brain parenchyma was observed in addition to granular cells (Fig. 6g). Normal brain histomorphology is present in the group of healthy mice (Fig. 6g). Pathohistological score of brain damage was significantly reduced in 4T1+ MSC14d when compared with 4T1+ PBS14d (P < 0.001; Fig. 6c). Also, in 4T1+PBS group, there was significantly higher brain damage than in 4T1+MSC14d treated group of animals (P < 0.001; Fig. 6c). Moreover, isolated primary tumors were pathohistologically examined (Fig. 6d). In the group of mice with 4T1+ MSC14d, the lymphocytic infiltration was more prominent and the necrosis fields were larger. In the 4T1+PBS14d group of animals, tumors were composed of malignant cells with polymorphic, hypertrophic nuclei, many nuclei, and no observable necrosis areas, indicating that the 4T1+PBS14d mice group has the greatest tumor damage, whereas 4T1+MSC14d group of mice exhibit significantly less tumor damage (P < 0.001; Fig. 6d). It was demonstrated that there was no significant difference in number of lung metastases between groups of 4T1+MSC14d and 4T1+PBS 14d (P > 0.05; Fig. 6e). There was no statistically significant difference between the 4T1+PBS14d group and the 4T1+MSC14d groups for the number of liver metastases, although the higher number of liver metastases was noticed in the 4T1+PBS 14d group (P > 0.05; Fig. 6f). Serum samples from 4T1+MSC14d-treated mice exhibited significantly elevated levels of vascular endothelial growth factor (VEGF) and TGF-β (P < 0.05; Fig. 6h, i), suggesting that MSCs administered during the advanced phase of breast cancer progression inhibited antitumor immunity by enhancing the production of proangiogenic and immunosuppressive cytokines in tumor-bearing mice.
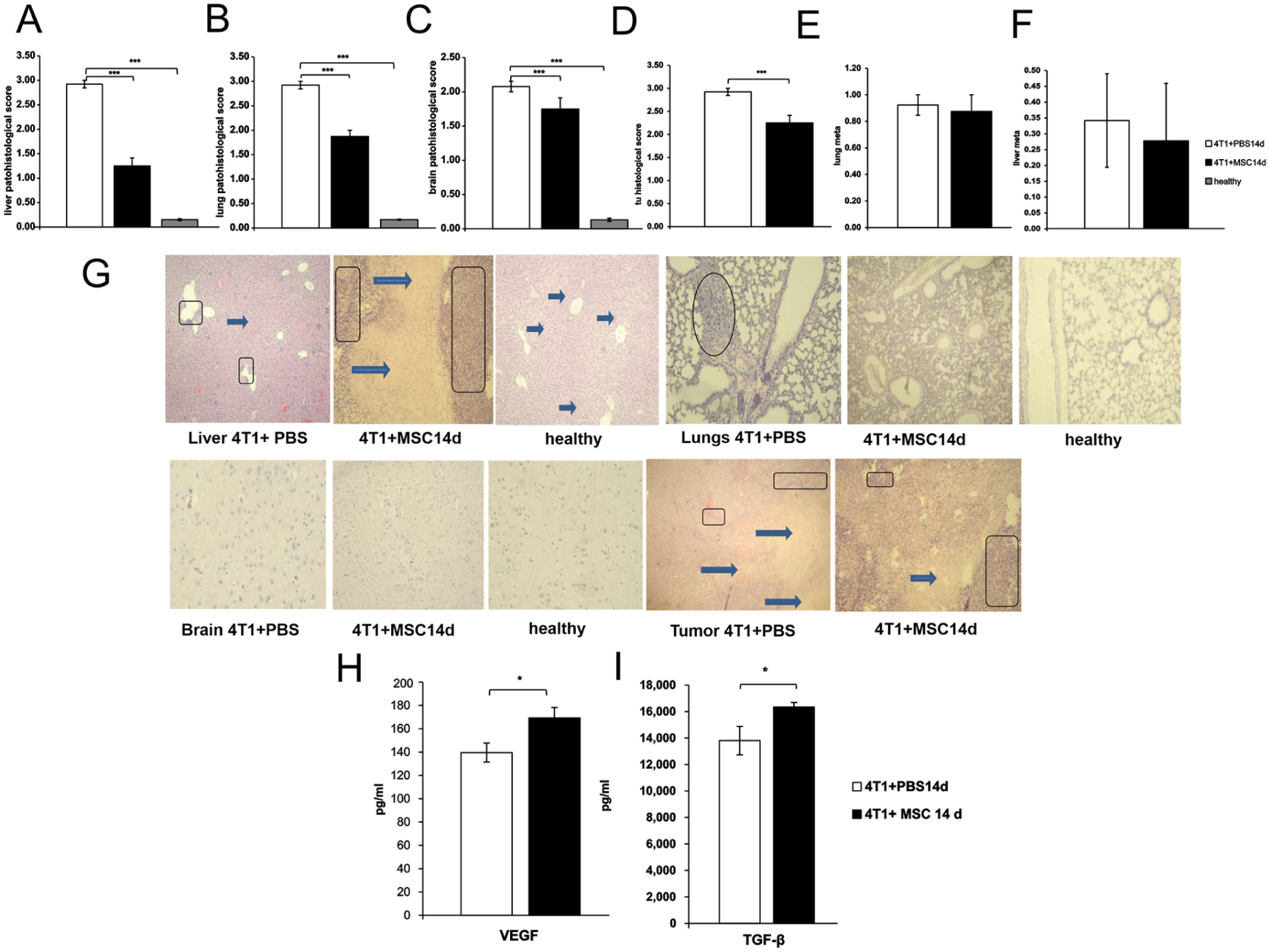
The progression of breast cancer is influenced by the timing of MSCs administration. Liver, lung, brain, and tumor pathohistological scores (a-d). Lung and liver metastases (e-f). Light-microscopic pictures through liver, lungs, brain, and tumors (H&E staining; magnification ×40): circle denote metastases, solid arrows signify necrosis, and rectangles represent lymphocyte infiltrates) (g). Serum samples from mice treated with 4T1+MSC14d had significantly higher amounts of proangiogenic and immunosuppressive cytokines VEGF (h) and TGF-β (i), compared to serum samples from mice treated with 4T1+PBS14d. Values shown as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
MSCs administered 14 days post-4T1 induction enhanced tumor progression by diminishing the antigen-presenting capabilities of tumor-infiltrating DCs and macrophages as well as impairing the cytotoxic functions of NK cells and T lymphocytes
In comparison to the tumors of 4T1+PBS14d treated mice, a markedly reduced percentage of innate immune cells, which are crucial for antitumor immunity (including cytotoxic NK cells, macrophages, and DCs), was noted in the tumors of 4T1+MSC14d-treated mice.
Mesenchymal stem cells, administered 14 days post-4T1 breast cancer induction, reduced the tumoricidal efficacy of NK cells and macrophages. This was demonstrated by a significantly lower percentage of tumor-infiltrating NK1.1.+ cells (P < 0.05; Fig. 7a), as well as decreased levels of IFN-γ (P < 0.05; Fig. 7b) and IL-17-producing NK1.1. cells (P < 0.05; Fig. 7c). The markedly reduced percentage of tumor-infiltrating F4/80+ macrophages (P < 0.05; Fig. 7d), along with DCs (P < 0.05; Fig. 7e), which include CD80-expressing (P < 0.05; Fig. 7f) and IL-12-producing DCs (P < 0.05; Fig. 7g), suggests that MSCs diminished the capacity of antigen-presenting cells to optimally activate T-cell-mediated antitumor immune responses.
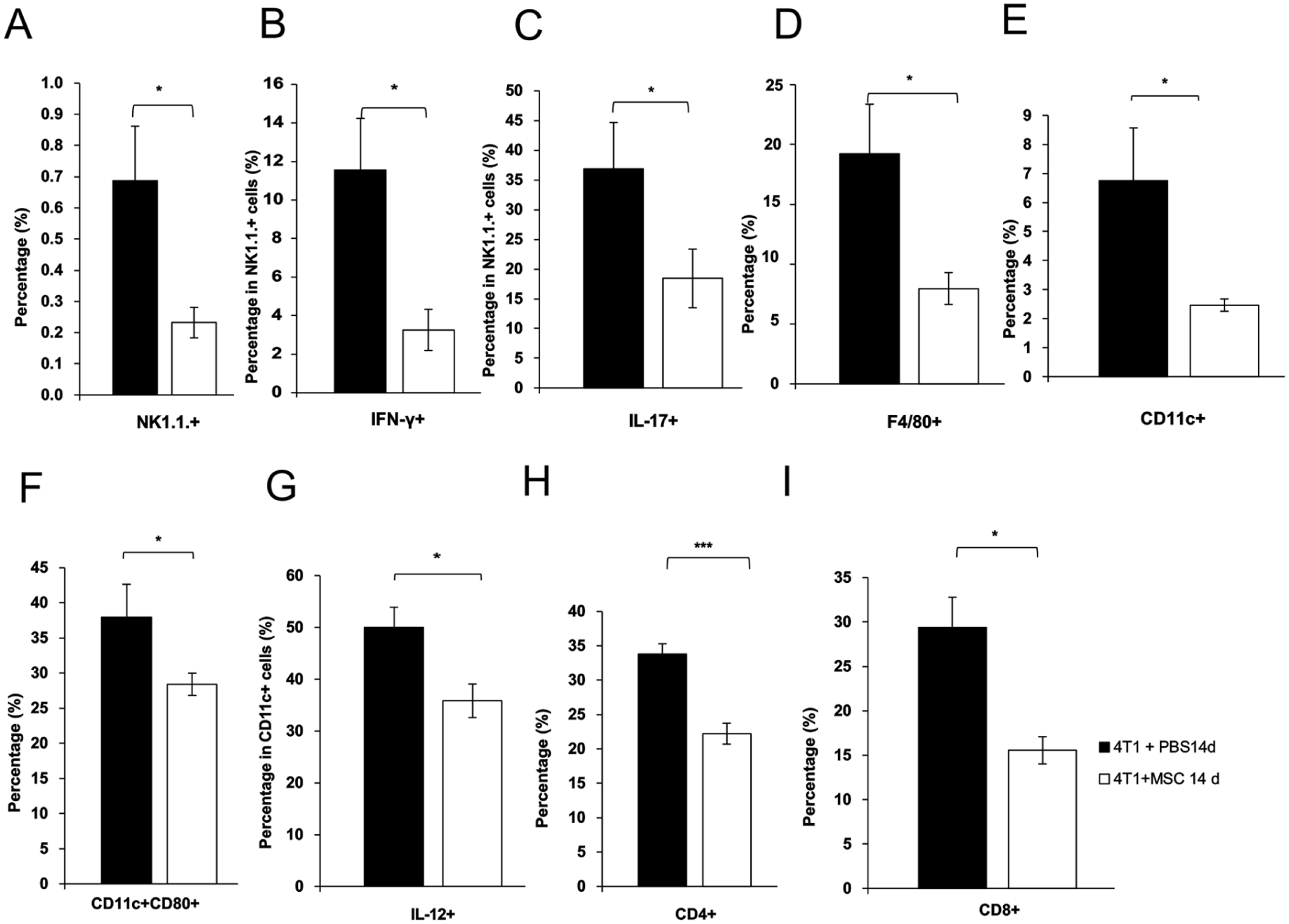
MSCs administered 14 days post- 4T1 tumor induction, facilitated tumor growth by inhibiting the cytotoxic activity of natural killer (NK) cells, diminishing the antigen-presenting capabilities of tumor-infiltrating macrophages and DCs, and suppressing T lymphocyte function. Significantly lower percentage of NK1.1.+ cells (a), IFN-γ and IL-17-producing NK1.1. cells (b, c), as well as F4/80+ macrophages (d), dendritic cells DCs (e) that are CD80- expressing (f), and IL-12- producing DCs (g), as well as CD4+T helper cells (h) and CD8+CTLs (i) were observed in the tumors of 4T1+MSC14d-treated mice compared to 4T1 +PBS14d-treated mice. Values shown as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
Fig. 7 illustrates that MSCs administered 14 days post-4T1 induction diminished the tumoricidal efficacy of CD4+Th1 and CD8+CTL cells. The percentages of both subpopulations of effector T lymphocytes, CD4+ T helper cells (P < 0.001; Fig. 7h) and CD8+ CTLs (P < 0.05; Fig. 7i), were markedly diminished in the breast tumors of mice treated with 4T1+MSC14d relative to those treated with 4T1+PBS14d.
Administration of mMSCs 14 days after 4T1 induction significantly altered the cellular composition of the spleen in mice, by increasing the percentage of regulatory CD3 and CD4 T cells thereby facilitating tumor progression and growth
The spleens of 4T1+MSC14d exhibited a significantly elevated percentage of CD25-, CD25+FoxP3- expressing CD3+cells (P < 0.05; Fig. 8a; P < 0.001; Fig. 8b, c), as well as significant expansion of Tr1-like (Type 1 regulatory T) cell populations characterized as FoxP3⁻ CD4⁺ cells producing TGF-β and IL-10 (Fig. 8d, e). Furthermore, there was an increased percentage of CD25-, FoxP3-, and CD25+FoxP3- expressing CD4+ cells (P < 0.05; Fig. 8f, P < 0.001; Fig. 8g–i) and CD25+TGF-β producing CD4+cells (P < 0.05; Fig. 8j) when compared to the corresponding percentages in the spleens of mice from the 4T1+PBS14d group. This indicates that MSCs administration 14 days post-4T1 induction augmented immunosuppressive capacity, thereby contributing to enhanced tumor growth as well as progression.

The administration of mMSCs 14 days post-4T1 induction significantly elevated the percentage of regulatory CD3 and CD4 T cells, thereby promoting tumor development and growth. Significantly elevated percentage of CD25- and CD25+FoxP3- expressing CD3+ cells (a–c), as well as TGF-β and IL-10-producing FoxP3⁻ CD4⁺ cells in 4T1+MSC14d group of mice (d, e) when compared to the 4T1+PBS 14d group of animals. Also, there was an increased percentage of CD25-, FoxP3-, and CD25+FoxP3- expressing CD4+ cells (f, g, h and i) and CD25+TGF-β producing CD4+ cells in 4T1+MSC14d when compared to 4T1+PBS 14d group of animals (j). Values are presented as the mean ± SEM; n = 9 mice per group; *P < 0.05, ***P < 0.001.
Discussion
Mesenchymal stem cells are known for their ability to home to tumor sites, but their role in cancer progression is highly context-dependent. Previous studies have shown that MSCs can exert both anti-tumor and pro-tumor effects, depending on the stage of cancer, the microenvironment, and the specific conditions under which they are administered11,27. Our study contributes to this understanding by demonstrating the complex, dual role of MSCs in the 4T1 breast cancer model. In our experiments, the timing of MSCs administration played a critical role in determining their effects on tumor progression. Early injection of MSCs, 1 day after 4T1 tumor induction, resulted in significant inhibition of tumor growth and metastasis. This was accompanied by increased survival, suggesting that MSCs can enhance the phenotypes consistent with antitumor potential when administered at the initial stages of cancer development. The effects of early MSCs treatment were associated with an increase in cytotoxic immune cells, such as NK cells and CD8+ T cells, and a shift toward a pro-inflammatory cytokine profile, with elevated levels of IFN-γ and TNF-α. These findings are consistent with our previous work, which showed that early MSCs treatment can boost the immune system’s ability to target and eliminate tumor cells 28 . However, the outcomes were markedly different when MSCs were administered at a later stage of tumor progression, 14 days post-4T1 induction. In this case, MSCs promoted tumor growth and metastasis, with a reduction in survival observed in treated animals. The late administration of MSCs led to a shift toward an immunosuppressive environment, characterized by an increase in regulatory T cells (Tregs) and the secretion of immunosuppressive cytokines such as IL-10 and TGF-β. This finding supports the notion that MSCs, when introduced at advanced stages of cancer, can contribute to immune evasion and promote tumor progression by dampening the immune response. The dual roles of MSCs observed in this study are further emphasized by their ability to modulate the tumor microenvironment in a manner that is highly dependent on their timing of administration. Early MSCs treatment enhanced the infiltration of antigen-presenting cells such as DCs and macrophages, which are crucial for initiating adaptive immune responses. Additionally, MSCs administered at this stage also induced the activation of cytotoxic T cells and NK cells, thereby promoting tumor surveillance and immune-mediated tumor cell death. Conversely, late MSCs treatment reduced the presence of these immune cells within the tumor and spleen, favoring the expansion of Tregs, which are known to suppress anti-tumor immunity. The role of cytokines in these responses cannot be overlooked. Our study demonstrated that early MSCs treatment led to an increase in pro-inflammatory cytokines like IFN-γ and TNF-α, which are key drivers of anti-tumor immunity. On the other hand, late MSCs administration resulted in the upregulation of immunosuppressive cytokines, including IL-10 and TGF-β, which are linked to tumor progression and metastasis. This suggests that the inflammatory microenvironment in which MSCs are introduced plays a pivotal role in determining their function. When MSCs are exposed to a pro-inflammatory environment, they appear to promote anti-tumor immunity, whereas in an immunosuppressive setting, they may contribute to tumor growth. Our findings are in line with several other studies that have reported stage-dependent effects of MSCs in different tumor models. In particular, research on the 4T1 breast cancer model has highlighted the importance of MSCs timing, as Zheng et al. 29 showed that MSCs administered at different sites (local vs. distant) had opposing effects on tumor growth and immune response. Additionally, Jazdeje et al. 30 observed that the administration of MSCs at various time points could either enhance or suppress immune responses, further reinforcing the need for careful consideration of the timing and method of MSCs administration in cancer therapies. In contrast to the pro-tumor effects seen with late MSCs treatment, our study indicates that early MSCs administration can improve animal survival by augmenting the immune system’s capacity to control tumor growth. However, the benefits of early MSCs treatment were not without limitations. While tumor growth was reduced, animal weight was lower in the MSCs-treated group, likely due to the reduced tumor size, but this did not reach statistical significance in comparison to the control group. Moreover, while early MSCs administration reduced metastasis to the lungs and liver, the extent of these reductions was not always statistically significant, indicating that further optimization of treatment parameters may be necessary to achieve more pronounced effects on metastasis. The complexity of MSCs’ role in cancer is underscored by the variation in outcomes across studies, with factors such as the source of MSCs, the route of administration, the tumor type, and the timing of treatment all influencing the results21,29–31. Our study adds to this body of evidence by showing that the effects of MSCs on tumor progression and metastasis are highly dependent on the timing of their administration. Early MSCs treatment appears to promote anti-tumor immunity, while late treatment may enhance tumor progression through immune suppression.
Our results suggest that the MSCs “licensing” phenomenon is critical for their divergent effects32,33. When MSCs engraft in tissue characterized by low levels of inflammatory cytokines, they adopt a pro-inflammatory phenotype (MSC1), thereby acquiring the ability to elicit a robust inflammatory response32,34,35. Conversely, MSCs subjected to elevated levels of inflammatory cytokines (inflammatory licensing) adopt an immunosuppressive phenotype (MSC2), generate significant quantities of anti-inflammatory factors including IDO and PGE₂, and suppress immune responses32,34,35. Hence, in the early phase of tumor development, before establishment of a strongly inflammatory microenvironment, MSCs may retain their capacity to produce pro-inflammatory mediators and exert anti-tumor effects28,32,36. Conversely, as tumors progress and develop chronic inflammatory conditions with elevated pro-inflammatory cytokines, MSCs become increasingly licensed toward production of IDO, PGE₂, and other immunosuppressive molecules that facilitate tumor immune evasion33,37.
We selected the IP route of MSCs administration to potentially avoid the pulmonary first-pass effect that occurs with intravenous delivery, where up to 80% of cells become trapped in lung capillaries38–40. While IP administration has been used in various tumor models41–45, direct comparative studies examining IP versus IV efficacy in the 4T1 model remain limited28,30. The dose of 5 × 105 MSCs was selected based on commonly used ranges in murine studies, although optimal dosing requires further investigation through systematic dose-escalation studies28,30,39,46–49. Jazedje et al. 30 used 106 cells in the same 4T1 model and observed therapeutic effects with IP administration 30 . Bazhanov et al. 50 showed IP MSCs form functional aggregates with immune cells in the peritoneal cavity, and Wang et al. 51 and Giri and Galipeau 52 showed superior therapeutic efficacy of IP vs. IV delivery51,52. A single dose administration was employed to isolate the temporal effects of MSCs application from potential confounding variables associated with repeated dosing regimens. Future studies should include direct head-to-head comparisons of administration routes (IP vs IV vs intratumoral), comprehensive dose-response analyses, and evaluation of multiple-dose regimens to optimize MSC-based therapeutic strategies for different clinical scenarios (early intervention vs established disease)38,39,51,52.
Although we observed robust changes in the anti-tumor immunological profile (increased tumor-infiltrating NK cells, DCs, and cytotoxic T cells expressing FasL and Granzyme B), tumor volume reduction was more modest (30%-40% at day 35). This apparent discrepancy should be interpreted in the context of the highly aggressive and immunosuppressive nature of the 4T1 model53–55, administration of a single MSC dose, and complex mechanisms of tumor immune evasion including PD-L1 expression and MDSC recruitment56–58. Similar patterns of robust immunological activation with modest tumor size reduction have been documented in other immunotherapeutic interventions in the 4T1 model59–61, reflecting the challenges of achieving complete tumor eradication in this aggressive model59,60. Importantly, the 30% to 40% tumor growth inhibition we observed is clinically meaningful and comparable to effects achieved with other immunomodulatory monotherapies in this model59,60. Combination of early MSC administration with other immunotherapeutic modalities (eg, checkpoint inhibitors) or administration of multiple MSC doses could potentially enhance anti-tumor efficacy and achieve greater tumor regression.
Finally, it should be noted that our study use a single tumor model (4T1). However, our findings are consistent with previous studies demonstrating similar timing-dependent effects of MSCs on anti-tumor immunity in other tumor models. Miloradovic et al. 28 showed that in the B16F10 melanoma model, MSCs applied on day 1 exhibited anti-tumor effects while MSCs on day 14 promoted tumor progression, which is an identical pattern to our current 4T1 study. Similarly, Hu et al. 44 demonstrated that early MSCs administration inhibited while late administration promoted progression in a colitis-associated colon cancer model. This consistency across three different tumor models (breast cancer, melanoma, and colon cancer) from independent research groups increases confidence in the general applicability of timing-dependent MSCs effects. Nevertheless, future studies examining additional tumor models of different tissue origins (eg, lung carcinoma and pancreatic cancer) would further confirm the universality of this phenomenon and identify potential tumor-type-specific variations. Eventually, it must be emphasized that although these data indicate timing-dependent immunomodulatory effects of MSCs administration in this experimental setting, extrapolation to human MSC-based therapies should be undertaken cautiously and will require validation in additional clinically relevant models and, ultimately, in appropriately designed clinical studies.
Study limitations
Several important limitations should be considered when interpreting our findings. First, our study is limited to a single murine tumor model (4T1 mammary carcinoma), which represents a highly aggressive, triple-negative breast cancer phenotype. The generalizability of our timing-dependent MSC effects to other tumor types (melanoma, colorectal cancer, lung cancer, glioblastoma) or even to different breast cancer subtypes (hormone receptor-positive, HER2-enriched) remains uncertain and requires systematic validation across multiple tumor models.
Second, we utilized only murine bone marrow-derived MSCs from a single commercial source. Mesenchymal stem cells derived from different tissue sources (adipose tissue, umbilical cord Wharton’s jelly, dental pulp) exhibit distinct immunomodulatory capacities, cytokine secretion profiles 62 . Furthermore, significant biological differences exist between murine and human MSCs in terms of surface marker expression, growth kinetics, and immunomodulatory mechanisms35,63. Therefore, our findings cannot be directly extrapolated to human MSC-based therapies without extensive validation using human-derived cells in appropriate preclinical models.
Moreover, our experimental endpoint was limited to 35 days post-tumor induction, which may not adequately capture long-term outcomes including tumor recurrence, late-stage metastatic complications, immune memory development, or chronic organ toxicity. Extended observation periods (60-90 days or lifetime studies) would be necessary to fully assess the durability of early MSC-induced anti-tumor immunity and to evaluate potential delayed adverse effects.
Additionally, murine tumor biology and 4T1 cells differ significantly from human pathophysiology, with 4T1 tumors showing poor immunogenicity and weak sensitivity to immunotherapies64–66. In addition, differences in cellular composition, immune cell trafficking, and tumor microenvironment architecture affect therapeutic efficacy, making direct extrapolation to human cancer patients challenging. Translation to clinical applications requires extensive preclinical validation using patient-derived xenograft models with human MSCs, followed by carefully designed early-phase clinical trials.
Moreover, our statistical analysis did not apply corrections for multiple comparisons. While our immune markers were selected a priori based on previous literature28,67,68, we acknowledge this as a limitation and that future validation studies with larger sample sizes would benefit from more conservative statistical approaches.
Although our study did not include direct in vitro cytolytic assays (such as 51Cr-release or CTL-tumor cell co-culture experiments), the significant increase in expression of validated cytolytic molecules (FasL and Granzyme B) in tumor-infiltrating CD8+ T cells and NK cells69–71, combined with histological evidence of tumor necrosis and reduced tumor growth, provide strong indirect evidence of enhanced anti-tumor cytolytic activity. Granzyme B expression has been extensively validated as a reliable surrogate of functional cytotoxicity in CTLs and NK cells in vivo70–73. Of note, Granzyme B and FasL expression have been extensively validated in the literature as surrogate markers of in vivo cytolytic function74,75. Importantly, the ultimate measure of effective cytolytic activity is tumor cell death and reduced tumor burden, both of which were significantly demonstrated in our MSC1d group through reduced tumor volume (P < 0.05), decreased metastasis (P < 0.05), histological evidence of central tumor necrosis, and prolonged survival76–79. Nevertheless, future studies incorporating direct functional cytotoxicity assays would provide additional mechanistic confirmation of enhanced tumor cell killing capacity.
Our study did not include direct MSC tracking (eg, fluorescent or bioluminescent labeling and genetic fate-mapping), which prevents precise assessment of MSC localization, persistence, and potential integration into tumor stroma. While our study employed IP administration to potentially avoid the pulmonary first-pass effect documented with intravenous delivery, the transient nature of MSC presence and predominant reliance on paracrine mechanisms remain consistent across administration routes. While IP administration may partially circumvent the pulmonary first-pass effect observed with IV delivery, MSCs administered via either route exhibit transient tissue presence and depend predominantly on paracrine mechanisms for their immunomodulatory effects51,80,81. However, extensive literature demonstrates that after systemic (IP or IV) administration, MSCs exhibit specific biodistribution patterns: they predominantly localize first to the lungs (where many become trapped in capillary beds), followed by distribution to tumor tissue, spleen, and other organs38,39,51,82. Viable MSCs persist in tissues for limited periods (eg, up to 24 h in lungs after IV administration), with cell debris subsequently cleared by phagocytes39,83,84. Rather than stably engrafting as structural cells in tumor stroma85–87. Mesenchymal stem cells exert their immunomodulatory effects primarily through paracrine signaling mechanisms including secretion of cytokines, chemokines, growth factors, and EVs that modulate surrounding immune and tumor cells84,88. Furthermore, Kidd et al. 86 specifically demonstrated tumor tropism after IP injection in an ovarian model. Finally, the functional consequences of MSC-immune cell interactions observed in this study (increased cytotoxic NK/CD8+ cell infiltration with early administration, Treg expansion with late administration) provide indirect evidence that MSCs have localized to the tumor microenvironment during the treatment window, even without direct cellular tracking. The temporal correlation between MSC administration timing and divergent immune outcomes provides functional evidence of MSC-immune interaction. Also, the pro-inflammatory cytokine profile observed only after early MSC administration, and the immunosuppressive profile observed only after late administration, are inconsistent with random MSCs biodistribution or passive systemic effects, and instead suggest active, context-dependent MSCs engagement with the tumor microenvironment. The transient nature of MSC presence combined with long-lasting effects on tumor immunity suggests that MSCs act as immunomodulatory catalysts that alter the tumor microenvironment during their brief presence, with sustained downstream effects mediated by the reprogrammed immune cells39,84. Future studies incorporating MSC tracking technologies would provide valuable mechanistic insights into the spatiotemporal dynamics of MSC-mediated immunomodulation and help determine whether differential MSCs localization patterns contribute to timing-dependent effects.
Finally, it is of paramount importance to acknowledge that IP delivery has its limitations compared to IV or intratumoral delivery. Following intravenous administration, MSCs show an initial accumulation in the lungs with later redistribution mainly to the liver, spleen, and kidneys, as summarized in biodistribution studies 89 . Furthermore, injection into body cavities such as the peritoneum is associated with limited systemic biodistribution, with cells distributing mainly to tissues in contact with the cavity fluid 89 . Consistent with this, Bazhanov et al. 50 showed that intraperitoneally delivered MSCs rapidly form aggregates with host immune cells, attach to peritoneal organs such as the omentum and mesentery, and only very small numbers of live cells are detected in systemic sites like spleen or lymph nodes, suggesting restricted access to systemic circulation. Finally, we acknowledge that direct delivery routes can yield different therapeutic profiles, as Seo et al. 90 demonstrated that the injection route significantly affected antitumor efficacy of cytokine-expressing MSCs, with intratumoral administration producing stronger tumor-specific T-cell responses and antitumor effects than intravenous administration in their models.
Future directions
Future studies should employ temporal secretome and transcriptomic profiling (proteomics, scRNA-seq)89–91 to characterize the transition from early pro-inflammatory MSCs phenotype to late immunosuppressive phenotype across tumor progression, with focus on IDO/PGE2 licensing mechanisms and EV cargo dynamics92–96. Early MSC administration (MSC1d) should be tested in perioperative combination strategies with immune checkpoint inhibitors 97 , chemotherapy, and radiation therapy to enhance early-stage anti-tumor immunity 98 . Conversely, late-stage disease warrants strategies to block endogenous tumor-associated MSCs through IDO1/COX-2/TGF-β pathway inhibition, chemokine-mediated MSCs recruitment blockade (CCR2/CXCR4 antagonists), and engineering of MSCs as therapeutic vectors carrying oncolytic viruses or pro-apoptotic agents99–101. Priority experiments include (1) short-term temporal secretome profiling and validation of MSC1d + anti-PD-1 combination efficacy; (2) medium-term optimization of MSCs delivery routes and biomarker development for MSCs phenotype prediction; and (3) long-term development of Good Manufacturing Practice (GMP)-grade MSCs manufacturing and clinical trials incorporating MSCs timing-dependent approaches for early-stage disease alongside MSC-blocking strategies for advanced malignancy.
Longitudinal tumor growth was evaluated using pairwise tests at individual time points rather than a unified repeated-measures model, which can forgo efficiency and inflate type I error across time. Future work will implement repeated-measures ANOVA and/or mixed-effects models to analyze tumor growth trajectories while accounting for within-mouse correlation91,102.
Conclusion
In conclusion, MSCs possess a context-dependent dual role in cancer therapy. Our findings emphasize the critical importance of timing in MSCs-based therapies. Early MSCs administration may offer a beneficial approach for enhancing anti-tumor immunity, while late MSCs administration may hinder immune responses and promote tumor growth. These results highlight the need for careful consideration of MSCs treatment protocols in clinical applications, as their effects on tumor progression can vary significantly depending on when they are introduced into the cancerous environment. Further research is needed to fully understand the mechanisms underlying these stage-dependent effects and to optimize MSCs-based therapies for cancer treatment.
Footnotes
Acknowledgements
The authors are grateful to Milica Dimitrijevic Stojanovic who assisted in the histological analysis of tissue samples.
Ethical Considerations
All experimental procedures were conducted in accordance with the Principles of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, 1985). Animal experiments were approved by the Ethical Committee for the Protection of the Welfare of Experimental Animals of the Faculty of Medical Sciences, University of Kragujevac (approval number: 01-2247; date of approval: 28th of May 2021).
Author Contributions
VV conceived the study; DPap, DPav, and DN conducted the experiments; VJ performed the Flow cytometry analysis; DPap, DPav, and DN wrote the manuscript. VV supervised the study and provided critical feedback and revisions. All authors read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Serbian Ministry of Science (grant no. 451-03-47/2023-01/200111) and Faculty of Medical Sciences University of Kragujevac (grant no. MP01/18).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data generated or analyzed in this study are included in this published manuscript.
Statement of Human and Animal Rights
This article does not contain any studies with human subjects, and animal subjects were handled in strict accordance with aforementioned protocols.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.