
Abstract
Several reports have been published on the isolation, culture, and identification of mesenchymal stem cells (MSCs) from different anatomical regions of the umbilical cord (UC). UC is suitable for standardizing methods of MSC isolation because it is a uniform source with high MSC numbers. Although the UC is considered a medical waste after childbirth, ethical issues for its use must be considered. An increased demand for MSCs in regenerative medicine has made scientists prioritize the development of MSC isolation methods. Several research groups are attempting to provide a large number of high-quality MSCs. In this study, we present a modulated explant/enzyme method (MEEM) to isolate the maximum number of MSCs from the entire UC. This method was established for the isolation of MSCs from different anatomical regions of the UC altogether. We could retrieve 6 to 10 million MSCs during 8 to 10 days of primary culture. After three passages, we could obtain 8–10 × 108 cells in 28–30 days. MSCs isolated by this method express CD73, CD90, CD105, and CD44, but they do not express hematopoietic markers CD34 and CD45 or the endothelial marker CD31. The genes SOX2, OCT4, and NANOG are expressed in isolated MSCs. The capacity of these MSCs to differentiate into adipocytes and osteocytes highlights their application in regenerative medicine. This method is simple, reproducible, and cost efficient. Moreover, this method is suitable for the production of a large number of high-quality MSCs from an UC in less than a month, to be used for cell therapy in an 80-kg person.
Introduction
Current research is increasingly focusing on mesenchymal stem cells (MSCs). The advantage of MSCs in cell therapy is their low immunogenicity. Therefore, they can be used as allogeneic cells (27). According to the International Society for Cellular Therapy (ISCT), different characteristics can be used for the identification of MSCs, such as adherence on a cell culture dish; expression of CD44, CD105, CD90, and CD73 surface markers; and differentiation into different cell lines, including adipocytes and osteocytes (12). MSCs can be isolated from different tissues, including adipose (7), cord blood (5,33), heart (5), lung (17), bone marrow (2), central nervous system (29), placenta (21), umbilical cord (UC) (28), and amniotic membrane (41) tissues. The advantages of MSCs over embryonic stem cells are their low immunogenicity, low tumorigenic potential, and few associated ethical and legal dilemmas (11). However, the low potential of differentiation and the variation in MSCs obtained from different tissues or even different regions of a tissue pose difficulties in standardizing methods for their isolation. MSCs can proliferate in large numbers and produce different molecules such as growth factors and immunomodulators. Therefore, they are considered suitable for cell therapy applications. In 1976, MSCs were isolated from bone marrow for the first time and were comprehensively studied (15,16). Although MSCs isolated from bone marrow have numerous applications in research and clinical trials, there are some problems in obtaining bone marrow samples, such as invasive sampling, limited number of donor volunteers, and reduced function of MSCs isolated from old persons. Therefore, researchers have been trying to find substitutes for bone marrow. Wharton's jelly, a mucoid tissue within the UC, is a good substitute for bone marrow since it is easily and abundantly available. Because the UC is considered medical waste after parturition, no legal issues are imposed. Furthermore, sampling and removing this tissue is not painful for the mother or child. MSCs isolated from Wharton's jelly easily proliferate and differentiate to different cell lines, and they are not tumorigenic (38). These cells have characteristics of MSCs, and they also express genes specific to embryonic stem cells, such as sex-determining region Y-box 2 (SOX2), octamer-binding transcription factor 4 (OCT4), and NANOG (26). Thus, MSCs isolated from the UC have more proliferation and differentiation potential than MSCs isolated from bone marrow or adipose tissue (14). Furthermore, the other advantages of the UC are the number of MSCs present and their chromosomal stability (24,40). Owing to the unique characteristics of UC-MSCs, there are several methods for their isolation and culture; however, there is no consensus on a standard method. Enzymatic digestion and explant culture methods are two ways of isolating MSCs from the UC. Various anatomical regions of the UC have different numbers of MSCs. Researchers have paid particular attention to two arteries and one vein of the UC, which contain MSCs in the subvascular regions. Wharton's jelly also contains MSCs (22). There are disagreements about the quality of MSCs isolated from different anatomical regions of the UC. Many researchers are attempting to find a standard method for isolating MSCs of the UC (18). In the present study, we introduce a rapid, simple, and effective method for isolating MSCs from the entire UC. We aimed at cost-effective isolation of a large number of MSCs in a short time. Once this method is standardized, it can be used in clinical applications, research, and cell banking.
Materials and Methods
Obtaining Informed Consent and Collecting UCs
Fifteen UCs were collected just after birth from normal, full-term newborns. The deliveries were performed by a gynecologist at Valiasr Hospital, Tehran University of Medical Sciences (TUMS). All experiments were approved by the Ethics and Clinical Studies Research Committee of TUMS according to Helsinki declaration. Informed consents were obtained from all mothers before surgery. Each UC was carried in a sterile dish containing phosphate-buffered saline (PBS; Life Technologies, Carlsbad, CA, USA), 100 U/ml penicillin (Sigma-Aldrich, St. Louis, MO, USA), and 100 U/ml streptomycin (Sigma-Aldrich) at 4°C and was immediately transferred to the Pediatric Urology Research Center of TUMS. The samples were placed in a refrigerator (4-8°C) for 1 to 18 h before processing.
Preparation of UCs and Vascular Isolation
The UCs were transferred to a biosafety cabinet II and were washed several times with sterilized PBS (Life Technologies) to remove traces of blood. Each UC was then placed in a 10-cm sterilized dish (Jet Biofil, Elgin, IL, USA) and was divided into 10-cm pieces. The blood vessels were carefully isolated from each piece of UC without any rupture. After isolation, both ends of each vessel were closed by a sterilized plastic clamp to prevent any cross contamination of endothelial and blood cells with MSCs. In this way, each piece of vessel formed a loop. The loops were mildly digested by collagenase type I (Life Technologies) (0.2 mg/ml) for 3 h at room temperature with mild shaking. Enzyme activity was neutralized by adding 10% fetal bovine serum (FBS; Life Technologies) in Dulbecco's modified Eagle's medium–low glucose (DMEM-LG; Life Technologies). The UC tissues were further cut into tiny pieces and digested with collagenase type I for 16 h (0.2 mg/ml; Life Technologies) Enzyme activity was neutralized by adding 10% FBS in DMEM-LG. In order to compare the results of those methods with the results of our modulated explant/enzyme method (MEEM), MSCs from each UC were isolated according to tissue explant and enzymatic digestion methods.
Culture of Total Length of UC by MEEM
After mild digestion (3 h in 0.2 mg/ml collagenase type I), vessel loops were placed in T75 flasks (SPL, Pocheon-si, South Korea) containing DMEM-LG supplemented with 15% FBS. After partial digestion (16 h in 0.2 mg/ml collagenase type I), the digested umbilical cord tissues were treated with PBS to decrease the viscosity and then centrifuged for 10 min at 500 × g. After removing the supernatant, the tiny partially digested tissues were transferred into the same T75 flasks containing the vascular loops. The UC pieces and the vascular loops were cultured in DMEM-LG and 15% FBS. Each UC was divided into three to six T75 flasks.
Time Taken by Cells to Form 90% Confluent Primary Culture in MEEM, Explant, and Enzymatic Methods
The UC tissues were grown in primary culture using MEEM, explant, and enzymatic methods in T75 flasks. For each of these three methods, the time taken to reach 90% cell confluency in primary culture was measured (Table 1). Obtained MSCs were seeded (1 × 104/well) in 24-well plates (Jet Biofil) in triplicate. Time taken to reach 90% cell confluency was calculated using an inverted microscope (TS100; Nikon, Gotenba, Japan). The number of cells was calculated using Neubauer slides. Cell viability was checked by trypan blue (Sigma-Aldrich) dye exclusion.
Time (Days) Taken to Reach 90% Cell Confluency for 15 Umbilical Cord Samples

MEEM, modulated explant/enzyme method. Average time until desired cell confluency attained was 9.2 days for MEEM, 20.7 days for explant method, and 20.1 days for enzyme method. The success rates were 100% (15/15), 60% (9/15), and 73.3% (11/15), respectively.
Growth Curve
For calculating the population doubling time (PDT), MSCs obtained by MEEM, explant, and enzymatic methods were plated in 24-well plates (Jet Biofil). The number of cells in each well was 4 × 104. Cells were counted daily in triplicates, and the mean was calculated for each method. Any increase in the number of cells was calculated for 7 consecutive days. Growth curves were plotted according to the number of cells. The PDT was calculated from the growth curves.
Immunophenotyping of UC-MSCs by Flow Cytometry
During passage 3 at 80–90% cell confluency, the medium was removed, and the cells were washed three times with warm PBS. They were then treated with 0.25% trypsin–EDTA (Life Technologies) for 3 min. The enzyme was neutralized by adding complete medium (DMEM-LG) and 15% FBS, and the cell suspension was centrifuged at 500 × g for 5 min. Cell viability was calculated by the trypan blue (Sigma-Aldrich) dye exclusion method. Next, the harvested cells were stained with CD105 (Cat: ab44967, 3 μg/106 cells; Abcam, Milton, Cambridge, UK), CD90 (Cat: ab23894, 1 μg/106 cells; Abcam), CD73 (Cat: ab54217, 2 μg/106 cells; Abcam), CD44 (Cat: ab6124, 1 μg/106 cells; Abcam), CD45 (Cat: ab10559, 3 μg/106 cells; Abcam), HLADR (Cat: ab8085, 0.01 μg/106 cells; Abcam), CD31 (Cat: ab9498, 50 μl/106 cells; Abcam), and CD34 (Cat: ab8536, 20 μl/106 cells; Abcam) anti-human-specific monoclonal antibodies. Negative control staining was performed by incubating cells with an identical concentration (as the concentration of the samples) of FITC- and PE-conjugated mouse IgG isotype antibodies. After 20 min of incubation at room temperature in the dark, stained cells were resuspended in 400 μl PBS and analyzed by CyFlow® Space flow cytometer (Partec, Münster, Germany). Histograms were generated based on computed results using Windows™-based flow cytometry software.
Evaluation of OCT3/4, SOX2, and NANOG Gene Expression by Reverse Transcription Polymerase Chain Reaction (PCR)
MSCs were collected in RLT buffer provided with RNeasy Plus Kit (Qiagen, Frederick, MD, USA) and stored at –80°C. Total RNA was extracted from MSCs using the RNeasy Plus Kit, and cDNA was generated. The PCR analysis was performed in all three methods using hot start Taq polymerase and a final volume of 25 μl per reaction. PCR was performed in a thermal cycler (Eppendorf Mastercycler Personal, Hamburg, Germany) to amplify target DNA according to the following protocol: initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 61°C for 30 s, and extension at 72°C for 60 s. The reaction was completed with a final extension at 72°C for 5 min. The success of PCR was tested with agarose (Sigma-Aldrich) gel electrophoresis, SYBER safe (Cat: S33102; Invitrogen, Waltham, MA, USA) DNA gel staining, and gel documentation (Gel Doc System; Nanolytik® NanoGel, Düsseldorf, Germany). The housekeeping gene, GAPDH was used as a positive PCR control. The gene-specific primers and size of PCR products are shown in Table 2.
Gene-Specific Primers, Size of Products, and NCBI Reference Sequence Numbers

Differentiation of UC-MSCs to Adipocytes
In all three methods, MSCs after the third passage were seeded (2 × 104 cells/well) in six-well plates (Jet Biofil) with complete medium (DMEM LG + 15% FBS). In 90% cell confluency, the complete medium was substituted by adipogenic medium containing 0.5 μM isobutyl xanthine (Sigma-Aldrich), 50 μM indomethacin (Sigma-Aldrich), 2.0 μM insulin (Sigma-Aldrich), 0.5 μM dexamethasone (Sigma-Aldrich), and 10% FBS. The medium was changed every 3 days. After 24 days, the cells were fixed by 4% paraformaldehyde (Sigma-Aldrich), washed with PBS, and stained with 0.3% Oil red O (Sigma-Aldrich).
Differentiation of UC-MSCs to Osteoblasts
In all three methods, MSCs after the third passage were seeded (2 × 104 cells/well) in six-well plates (Jet Biofil) with complete medium (DMEM LG + 15% FBS). In 80% cell confluency, the complete medium was substituted by osteogenic medium, containing 0.1 μM dexamethasone, 0.2 μM ascorbic acid 2-phosphate (Sigma-Aldrich), 10 mM glycerol 2-phosphate (Sigma-Aldrich), and 10% FBS. The medium was changed every 3 days. After 24 days, the cells were fixed using 4% paraformaldehyde (Sigma-Aldrich), washed with PBS, and stained with Alizarin red S (Cat: BI1009; Fluka, Buchs SG, Switzerland).
Results
Isolation and Culture of UC-MSCs by MEEM, Explant, and Enzymatic Methods
Umbilical cord primary cultures (N = 15) were obtained by MEEM, explant, and enzymatic methods. Based on calculated PDT and time to reach >90% cell confluency, the average of harvested MSCs per 10-cm piece of UC were estimated to be 550,000, 150,000, and 280,000 in MEEM, explant, and enzymatic methods, respectively. Using trypan blue dye exclusion, the mean percentages of viable cells were 90%, 95%, and 78%, respectively. Inverted microscopy revealed fibroblast-like morphology after 16 h for MEEM (Fig. 1A), after 48 h for the enzymatic method (Fig. 1B), and after 72 h for the explant method (Fig. 1C).

Fibroblast-like morphology in umbilical cord mesenchymal stem cell primary culture. (A) Modulated explant/enzyme method (MEEM) after 16 h. (B) Enzymatic method after 48 h. (C) Explant method after 72 h. Original magnification: ×4. Scale bars: 100 μm.
Time Taken to Reach 90% Cell Confluency and Success Rate in Primary Cultures
For all samples, the time required to reach 90% confluence was determined. The results are shown in Table 2. On an average, cells took 9.2, 20.7, and 20.1 days, respectively, to reach 90% confluency with MEEM, explant, and enzymatic methods. The respective success rates were 100% (15/15), 60% (9/15), and 73.3% (11/15).
Average Cell Counts and Culture Durations in Different Stages of MEEM Method
We isolated almost 30,000,000 MSCs from each UC in primary culture during the first 10 days (five T75 flasks each with 6,000,000 MSCs in 90% confluency). They were then subcultured every 6 days in the ratio 1 to 3. We had approximately 90,000,000 MSCs in day 16 (first passage at 90% confluency), 270,000,000 MSCs in day 23 (second passage at 90% confluency), and 810,000,000 MSCs in day 29 (third passage at the 90% confluency). We worked on 15 UCs during this project, and results of all the samples confirmed each other.
Growth Curve and PDT Estimation
Growth curve of UC-MSCs at passage 3 were plotted. PDT did not differ across MEEM, explant, or enzymatic methods (Fig. 2A). The PDT for the entire UC-derived MSCs was 36 h (Fig. 2B).
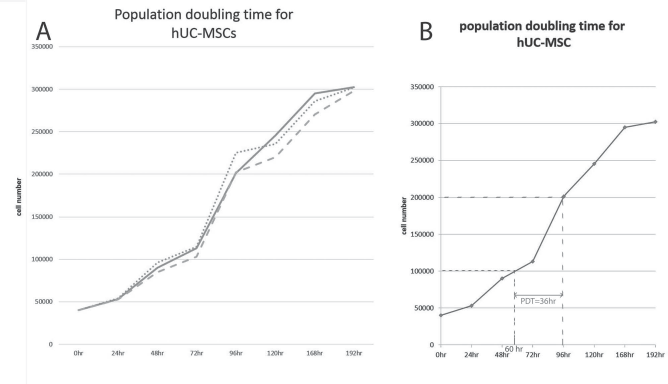
Growth kinetics of umbilical cord mesenchymal stem cells. Umbilical cord mesenchymal stem cells (40,000 cells per well) were plated in 24-well plates. Cells were counted in triplicate every day, and the mean number of cells was calculated for each method. (A) Triplicate MEEM (solid line), explant (dotted line), and enzymatic (dashed line) cultures were harvested for trypan blue dye exclusion cell count every day. (B) The population doubling time was calculated from the modulated explant/enzyme method (MEEM) curves.
Cell Surface Antigen Expression Profile of UC-MSCs by Flow Cytometry
UC-MSCs isolated by MEEM were positive for surface MSC antigens (CD105, CD90, CD73, and CD44) and negative for hematopoietic stem cell markers (CD45, CD34). These cells did not show expression of CD31 (endothelial cell marker) or HLADR (Fig. 3). The immunophenotype of total UC-MSCs obtained by MEEM was similar to those obtained by explant and enzymatic methods, which is in agreement with the findings of one study in 2006 (12) (data not shown).

Flow cytometry-based immunophenotyping of umbilical cord mesenchymal stem cells derived by MEEM. Cells are prominently positive for CD105, CD44, CD73, and CD90 and negative for HLADR, hematopoietic surface cell markers CD45, CD34, and endothelial cell surface antigen CD31.
Gene Expression Assessment by RT-PCR
To demonstrate the expression of pluripotent genes NANOG (Fig. 4A), SOX2 (Fig. 4B), and OCT4 (Fig. 4C) RT-PCR was performed on UC-MSCs obtained by MEEM, explant, and enzymatic methods (Fig. 4).

Gene expression by RT-PCR analysis of the umbilical cord-derived mesenchymal stem cells (UC-MSCs) obtained using modulated explant/enzyme method (MEEM), explant (EXP), and enzymatic (ENZ) methods. UC-MSCs express markers of embryonic stem cell transcription factors such as NANOG (A), SOX2 (B), and OCT4 (C). A 100-bp DNA ladder (Lad) was used as the DNA marker. A lymphoblastoid cell line was used as the negative control (NTC). An embryonic cell line (ESC) was used as the positive control (PTC).
Cell Culture Morphology Assessment of MEEM by Inverted Microscopy
After 16 h of MEEM culture, fibroblast-like attached cells appeared (Fig. 5A). After 48–72 h, small heterogeneous cell colonies were seen (Fig. 5B). The cells were fibroblast-like, flattened, and polygonal, with high nuclear/cytoplasm ratio. Seventy-two hours after the initiation of culture, vascular loops were removed carefully to avoid rupture. The cells and colonies increased markedly after 5 days (Fig. 5C), and flasks were confluent by about day 10 (Fig. 5D). DMEM-LG medium (2 ml) supplemented with 10% FBS was added every other day during MEEM culture of UC tissue without removing the previous medium.

Assessment cell culture morphology in MEEM by inverted microscopy. (A) After 16 h, (B) after 48–72 h, (C) after 5 days, (D) after 10 days. Scale bars: 100 μm.
Evaluation of Living Cells in Different Passages
Using trypan blue dye exclusion technique, the percentage of living cells obtained by MEEM for different passages was estimated to be about 97%.
Mesodermal Lineage Differentiation Potential of UC-MSCs Derived by MEEM
When placed in the specific differentiation media for 24 days, UC-MSCs obtained by MEEM, explant, and enzyme methods differentiated into osteocytes and adipocytes. No deposition of minerals or formation of lipid droplets was observed in the absence of differentiation medium (Fig. 6A–C). Deposition of calcium and mineralization of cells was observed by inverted microscopy as the appearance of red color after Alizarin red S staining (Fig. 6D, E, and F). Formation of lipid droplets and accumulation of lipid vacuoles was observed by inverted microscopy as the appearance of red color after Oil red O staining (Fig. 6G–I).

Osteogenic and adipogenic differentiation of UC-MSCs derived using the modulated explant/enzyme method (MEEM), explant (EXP), and enzymatic (ENZ) methods. (A, B, and C) Control; without the use of differentiation media. (D, E, and F) Alizarin red S staining after 24 days in osteogenic differentiation medium. (G, H, and I) Oil red O staining after 24 days in adipogenic differentiation medium. Scale bars: 50 μm.
Discussion
The MEEM method, we developed in this study, helped us isolate a larger number of MSCs from various anatomical regions of the UC in a shorter time than that required for enzymatic digestion and explant culture methods. Besides the advantages in ease of operation, MSC yield is high using this method. The isolated cells have an MSC-like morphology, express MSC-specific markers, and can differentiate into adipocytes and osteocytes.
MSCs can be isolated from different types of tissues (8,19) and have the potential for renewal and differentiating into different types of cells. Therefore, they are considered useful for various clinical applications. There is a need to review and optimize current techniques for isolating MSCs from the UC (9). In addition to the reproducibility of the technique, the morphology and phenotype of isolated cells should be maintained in different passages (26). Some groups have tried to optimize the use of enzyme methods. Improvement in the quality of the isolated cells among these attempts have been reported (18,35). The advantages of our study over others include isolation of larger number of MSCs from umbilical cord to shorten the length of the enzymatic and explant processes. Based on the evidence, MSCs isolated from different regions of the UC do not show similarities in number, differentiation, and proliferation (22,36). This underscores the importance of using the entire UC in isolating MSCs, which have uniform potential for proliferation and differentiation.
Umbilical cord can be considered biological waste. Hence, there are only a few minor ethical issues associated with the use of umbilical cord in regenerative medicine. The UC-MSCs have similar characteristics to embryonic stem cells because they are formed in the early stages of embryonic development (39). Although UC-MSCs are considered to be adult stem cells, the potential for differentiation is greater than that of MSCs derived from other sources (31). Owing to the above-mentioned features, researchers have tended to isolate MSCs from the umbilical cord to be used in regenerative medicine. Thus far, no standard isolation method for UC-MSCs is available. It has been reported that differences in isolating and culture methods can lead to differences in pluripotent potential and gene expression in cells (25). In addition, genetic and epigenetic changes of obtained cells can affect their clinical uses due to loss of plasticity (32). It should be noted that the expression of cell surface markers is influenced by factors secreted by accessory cells in primary culture (10). At present, there are enzymatic and tissue explant methods for isolating UC-MSCs. Cell yields by the explant method are low, and the enzymatic method is mostly variable, with occasional instances of cells with damaged membranes (37). In this study, we used a combination of two conventional protocols: tissue explant and enzymatic method (37). Characteristics of cells obtained by MEEM were compared with those obtained by conventional enzymatic and explant methods. The aim of this investigation was to introduce an efficient and reproducible method for isolating UC-MSCs. It seems that standard features of MEEM are higher than that of conventional methods.
We are the first to have cultured different parts of the UC, such as subvascular region, Wharton's jelly, and the subepithelial layer simultaneously using MEEM. We found that simultaneous culture of different regions of UC using MEEM accelerates the process of attaining desired cell confluency in primary culture. We did not observe any difference in the time taken to reach cell confluency after primary cultures using different methods. We believe the large quantities of cells obtained by MEEM in comparison with enzymatic and explant methods is one important cause of reduction in time to reach confluency in primary culture. The estimated number of isolated cells from 10-cm pieces of the UC using MEEM was about twice that obtained using the enzymatic method and four times than that obtained using the tissue explant method. Cell quality was the same for MEEM and explant methods, but cell quality for the enzymatic method was lower owing to membrane damage. The use of collagenase combined with trypsin and hyaluronidase can damage cell membranes, which reduces strength of cell adhesion and growth rate (6).
Wharton's jelly contains insoluble collagen, hyaluronic acid, and sulfated glycosaminoglycans around MSCs, all of which reduce the migration of cells from the tissues to flask surfaces. Using enzymes to dissolve these agents can facilitate the entry of cells into culture dishes. Different enzymes such as collagenase 1, 2, and 4, trypsin, and hyaluronidase were used alone or in combination. MSCs are very sensitive to trypsin, and other enzymes have destructive side effects (42). In MEEM, the tissues were finely sliced, and then type 1 collagenase was used for mild digestion of the entire UC. Before full digestion of UC tissues, enzyme activity was neutralized using complete medium. The entry of cells from mildly digested tiny tissues to the flask surface was facilitated by MEEM. Because of mild digestion, cell damage was minimal, and therefore cells had high adhesion strength and good growth potential. Observation of fibroblast-like cells after only 16 h indicates timely removal of enzymes and no damage to the cells. Owing to the comparable age of newborns, there is no considerable difference in UC-MSCs derived from different UCs. Thus, MEEM can be introduced as a standard method for isolating UC-MSCs. In the tissue explant method, the cells appeared on the surface of flasks very late due to low rate of release of cells. In MEEM, the rate of release of cells is higher, and the number of primary isolated cells is greater than that obtained by other methods. Furthermore, with regard to cell adhesion strength, expression of MSC surface markers, and mesodermal differentiation potential, the quality of the MSCs isolated was adequate. There were no differences in cell morphology between MSCs obtained by MEEM and conventional methods at passage 3. The isolation success rate was 100% in MEEM versus 73% in the enzymatic method and 60% in the explant method; this can be ascribed to the simplicity and reproducibility of MEEM. Application of MSCs in regenerative medicine requires a large number of high-quality cells. One of the advantages of MEEM is obtaining 8 × 108 ± 1.5 × 107 cells in less than a month. Considering the fact that the average number of cells required for bone marrow transplantation clinical trials is 1–2 × 106 MSCs per kg body weight (4), the cells obtained by MEEM during a month is sufficient for use in a patient weighing 80 kg.
Availability of MSC banks is one of the most challenging aspects in regenerative medicine. Assessment of growth kinetics of UC-MSCs, as well as existing MSC surface markers (CD90, CD 70, CD44, and CD 105), lack of hematopoietic (CD34, CD45) and endothelial (CD34) markers, and their multilineage differentiation potential, indicated that suitable and efficient cells were isolated using MEEM. Because growth factors are not needed for this method, the operational cost is reduced. Another advantage of our method is the easy availability of tissues since biological waste is being utilized.
Because of immunomodulatory properties of MSCs, their allogeneic use as universal donor cells is being considered (23). Some studies have shown that MSCs can be used for allogeneic purposes. For this, the use of serum (e.g., FBS) remains a drawback in our study. Some studies have recommended the use of autologous serum instead of FBS (13). However, we did not consider this in our present study. The increase in contact among MSCs (20) and the presence of autocrine and paracrine growth factors from different accessory cells (3) can accelerate achievement of cell confluency in primary culture in MEEM. Some researchers believe that homogeneous cells isolated from specific parts of the UC are most useful in regenerative medicine (30). We postulate that a large number of heterogeneous MSC populations obtained from different parts of the UC by MEEM can increase secretion of various relevant factors that leads to better proliferation and differentiation of MSCs. However, this theory warrants future research. The PDT of MSCs obtained by MEEM (36 h) is shorter than that of MSCs obtained from bone marrow (4 days) in previous studies (1). Because of this, there is less of a risk of chromosomal changes and cell abnormalities in these cells (34). In view of the above-mentioned issues, we propose the use of MEEM as an efficient and simple method for the standardization of UC-MSC isolation.
Footnotes
Acknowledgments
This study was supported by the Tehran University of Medical Science (TUMS) and Shahrekord University of Medical Science (SKUMS) (No. 18100). We thank the staff of the Pediatric Urology Research Center of TUMS, Cellular and Molecular Research Center of SKUMS, and Pathology and Oncopathology Research Centre of Iran University of Medical Science for their sincere assistance. None of the authors has direct or indirect commercial financial incentive associated with publishing the article. The authors declare no conflicts of interest.