
Abstract
BRCA1 mutation is closely linked to triple-negative breast cancer (TNBC), a subtype characterized by early onset, aggressive clinical course, and marked genomic instability. Although poly ADP ribose polymerase (PARP) inhibitors have expanded treatment options, resistance driven by cancer stem cells (CSCs) and incomplete interception of premalignant progression remain pressing challenges. In a recent study, Liu et al. developed a human induced pluripotent stem cell (iPSC)-based breast cancer model that follows BRCA1‑driven tumorigenesis from mammary differentiation to invasive disease and identify S100P as a BRCA1‑repressed effector associated with cancer stemness. These findings provide a human genetic context to probe early oncogenesis and nominate the S100P–RAGE axis as a precautionary—but promising—target for risk interception.
Recent advances in human disease modeling highlight the importance of preserving both the native genetic background and the developmental trajectory of the target tissue in cancer research. Liu et al. 1 apply this principle to breast cancer by establishing a human induced pluripotent stem cell (iPSC)-based platform that reconstructs the entire tumorigenic cascade initiated by a pathogenic BRCA1 mutation (Fig. 1). Peripheral blood monocytes from a BRCA1-mutation carrier were reprogrammed into iPSCs and directed through a rigorously defined mammary differentiation protocol, generating the first human-relevant model to recapitulate tumorigenesis from premalignant epithelium to fully penetrant, histologically validated tumors. Mechanistically, BRCA1 loss upregulates the calcium-binding protein S100P, which in turn activates a cancer stem cell (CSC) program via the RAGE/NF-κB axis—presenting a tractable molecular vulnerability for precision prevention and for eradicating residual disease following standard-of-care PARP inhibition. This work is a timely complement to the study by Weddle et al. 2 , which uses patient-derived iPSCs to evaluate PARP inhibitor sensitivity. Together, these studies position iPSC technology as a critical bridge between germline genetics and precision oncology.
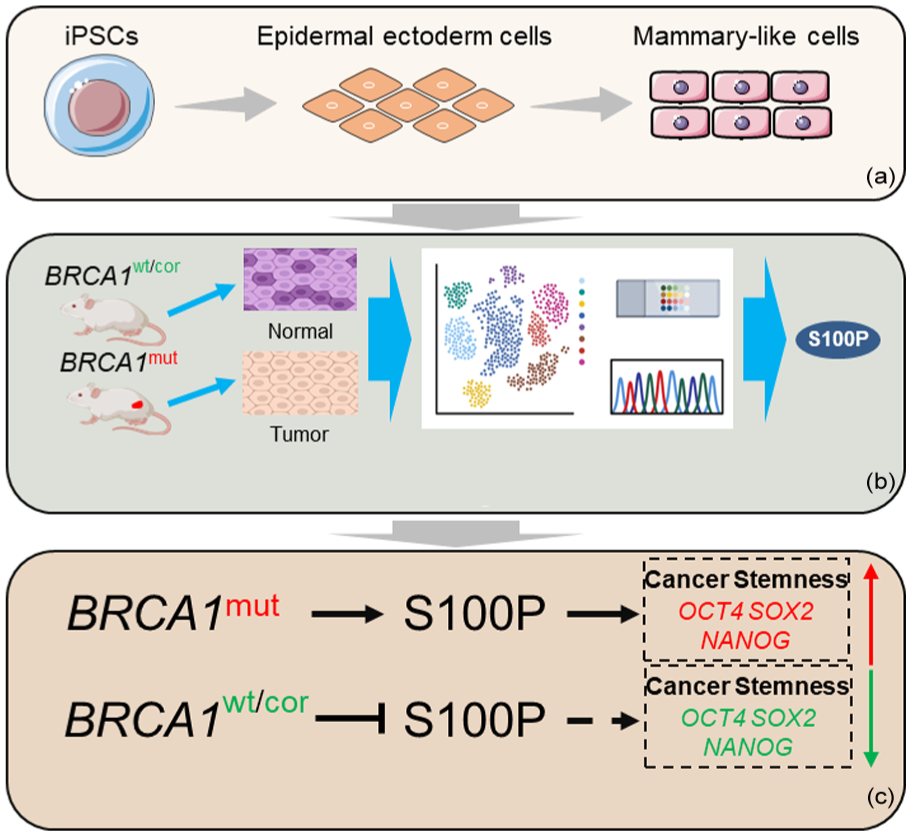
Human iPSC-derived mammary model recapitulates BRCA1-mutant tumorigenesis and reveals S100P-driven cancer stemness. (a) Establishment of method to generate iPSC-derived mammary cells. (b) iPSC-based model recapitulates BRCA1-mutant breast cancer tumorigenesis and gene signature. (c) iPSC-derived mammary model links BRCA1 mutation–driven tumorigenesis to S100P-mediated stemness.
The central advance of Liu et al.’s study (Fig. 1a) is a technically rigorous differentiation protocol that converts human iPSCs into functional mammary-like cells (iPSC-MCs). Guided by developmental biology principles, iPSCs are first directed toward an epidermal ectoderm fate and then differentiated into bipotent mammary epithelial cells co-expressing basal (CK5) and luminal (CK8) cytokeratins. These cells form branching organoids in three-dimensional culture and regenerate ductal outgrowths enriched in human leukocyte antigen (HLA)-positive epithelium when transplanted into cleared murine fat pads. This degree of functional maturation surpasses earlier approaches that relied on stochastic embryoid body formation and achieved only limited lineage fidelity3–5. By reaching a differentiation efficiency above 90%, Liu et al. overcome a long-standing bottleneck that had restricted human mammary differentiation to low-yield protocols.
Building on this platform, the authors introduced a BRCA1-null background by applying CRISPR/Cas9 to edit patient-derived iPSCs carrying a pathogenic BRCA1 mutation. Isogenic wild-type and gene-corrected lines served as stringent controls, enabling causal inference without the inter-individual variability that often complicates patient-derived xenograft studies (Fig. 1b). Upon transplantation, BRCA1-mutant iPSC-MCs produced tumors histologically indistinguishable from clinical TNBC—high-grade, triple-negative by receptor status, and displaying Ki67 and P120 staining patterns indicative of aggressive proliferation. Whole-exome sequencing of these tumors reproduced the mutational landscape of human BRCA1-mutant breast cancer, including the recurrent P53-R280K substitution and an elevated transversion signature characteristic of homologous recombination deficiency. In doing so, this model not only captures the initiating genetic lesion but also recapitulates the evolutionary pressures that drive subsequent genomic instability, positioning it as a powerful tool for dissecting early tumorigenic events in a human genetic context.
Beyond faithfully recapitulating disease pathology, the study’s key mechanistic contribution is the identification of S100P—a calcium-binding protein—as the molecular link between BRCA1 loss and CSC expansion. Integrative analyses demonstrated that BRCA1 directly binds and represses the S100P promoter; loss-of-function mutations in BRCA1 relieve this repression, driving S100P overexpression. Functional assays confirmed S100P’s essential role: knockdown markedly reduced tumor volume and decreased ALDH1⁺CD44⁺CD24⁻ CSC populations, whereas overexpression enhanced mammosphere formation and CSC marker expression. Clinically, elevated S100P expression correlated with poorer relapse-free survival (RFS) and distant metastasis-free survival (DMFS) in breast cancer cohorts, consistent with its established roles in promoting migration and therapy resistance.
Mechanistically, S100P engages RAGE to activate MAPK/ERK and NF-κB signaling pathways extensively characterized in cancer models6,7. In the BRCA1-deficient iPSC-based breast cancer model, S100P overexpression increased the levels of core pluripotency transcription factors (OCT4, SOX2, NANOG) and the breast CSC marker ALDH1, whereas S100P inhibition suppressed these stemness-associated features. These findings position the S100P–RAGE axis as a central regulator of tumor-initiating cells with significant therapeutic potential. In preclinical studies by Liu et al., the RAGE inhibitor FPS-ZM1 suppressed BRCA1-mutant tumor growth by disrupting S100P-driven stemness, suggesting a possible role in risk interception.
Considered alongside the work of Weddle et al., these findings acquire additional depth. Weddle’s team reprogrammed primary breast tumors from nine patients—spanning luminal, HER2-enriched, and triple-negative subtypes—into iPSCs and differentiated them into mammary epithelial cells (BC-hiPSC-MECs). While their focus was on drug responsiveness rather than tumor initiation, both studies share a commitment to preserving patient-specific genetics within an experimentally tractable system. Weddle et al. showed that BC-hiPSC-MECs harboring pathogenic BRCA1 or BRCA2 variants are highly sensitive to olaparib and talazoparib, confirming synthetic lethality in a human cellular context. Using CRISPR, they further demonstrated that the Ashkenazi founder mutation BRCA1 is both necessary and sufficient for PARP inhibitor hypersensitivity. Liu et al. extend this by showing that even in a BRCA1-null background, modulation of S100P can shift CSC dynamics and potentially influence drug tolerance. Together, these studies offer complementary perspectives: Weddle et al. illuminate the immediate therapeutic response, whereas Liu et al. uncover early oncogenic reprogramming events that may eventually contribute to resistance.
From a methodological standpoint, both groups addressed a long-standing limitation of iPSC technology—its perceived inefficiency in reprogramming solid tumors—by optimizing differentiation timing and media transitions. Liu et al. achieved over 90% differentiation of iPSCs into mammary epithelial cells, defined by CK8/CK5 co-expression, whereas Weddle et al. attained similarly high efficiencies using an optimized Sendai virus–based reprogramming approach. These refinements transform what was once a niche, technically demanding method into a robust platform suitable for both large-scale screening and personalized therapeutic development.
Extensive evidence indicates that traditional cancer cell lines, having undergone decades of in vitro adaptation, frequently lost the early molecular signatures of tumor initiation while acquired genetic drift and other artifacts 8 . Patient-derived organoids preserve native tissue architecture and cellular heterogeneity but often display variability and lack isogenic controls, complicating causal inference over developmental time 9 . By contrast, iPSC-based models capture germline predisposition within the relevant developmental context and, when coupled with gene editing, allow direct dissection of temporal causality in tumorigenesis 10 . These models occupy a unique niche: they enable researchers to observe the “first domino” in malignant transformation—how a single inherited mutation distorts lineage commitment, rewires transcriptional networks, and ultimately licenses neoplastic conversion. By capturing this sequence in human cells, Liu et al. provide a rational framework for intercepting tumorigenesis before it becomes clinically apparent.
Looking forward, several key questions emerge. First, does the BRCA1–S100P axis operate across all basal-like TNBCs, or is it restricted to germline BRCA1 carriers? BRCA1 promoter hypermethylation is common in sporadic TNBC and correlates with reduced BRCA1 expression and poor prognosis11–13, suggesting that epigenetic silencing could similarly modulate critical oncogenic pathways. However, large-scale TCGA analyses directly linking BRCA1 promoter methylation to S100P upregulation remain unavailable. Second, how does S100P-driven stemness intersect with immune-evasion mechanisms increasingly recognized in BRCA1-mutant tumors? Liu et al. note that RAGE signaling can influence cytokine secretion, hinting at a possible connection to the immune-rich microenvironment characteristic of many TNBCs. Finally, can S100P inhibition synergize with PARP inhibitors to delay or prevent therapeutic resistance? Preclinical studies combining PARP and RAGE inhibition in breast cancer may offer valuable translational insights.
In conclusion, Liu et al. present a landmark study that broadens the scope of iPSC technology from hematologic malignancies to solid tumors and from drug-response profiling to modeling oncogenic initiation. By delineating the BRCA1–S100P–RAGE circuitry in human mammary development (Fig. 1c), they provide both a mechanistic framework for understanding TNBC heterogeneity and a strategic roadmap for precision prevention. Viewed alongside Weddle et al.’s demonstration of patient-specific PARP inhibitor responses, this work positions iPSC-derived breast cancer models as a cornerstone of next-generation precision oncology.
Footnotes
Acknowledgements
We thank Ying Lou for providing insightful comments and suggestions.
Ethical considerations and consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contributions
G.Z. drafted and edited the manuscript. B.Z. critically revised the manuscript and approved the final version for submission.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Statement of human and animal rights
This article does not contain any studies with human or animal subjects.
Statement of informed consent
There are no human subjects in this article and informed consent is not applicable.