
Abstract
Stem cells have the potential to replace defective cells in several human diseases by depending on their self-renewal and differentiation capacities that are controlled by genes. Currently, exploring the regulation mechanism for stem cell capacities from the perspective of methyltransferase-like 3 (METTL3)-mediated N6-methyladenosine modification has obtained great advance, which functions by regulating target genes post-transcriptionally. However, reviews that interpret the regulatory network of METTL3 in stem cells are still lacking. In this review, we systematically analyze the available publications that report the role and mechanisms of METTL3 in stem cells, including embryonic stem cells, pluripotent stem cells, mesenchymal stem cells, and cancer stem cells. The analysis of such publications suggests that METTL3 controls stem cell fates and is indispensable for maintaining its normal capacities. However, its dysfunction induces various pathologies, particularly cancers. To sum up, this review suggests METTL3 as a key regulator for stem cell capacities, with further exploration potential in translational and clinical fields. In conclusion, this review promotes the understanding of how METTL3 functions in stem cells, which provides a valuable reference for further fundamental studies and clinical applications.
Keywords
Highlights
METTL3 maintains the normal renewal and differentiation of stem cells.
METTL3 is required for embryo formation and development.
METTL3 decides pluripotent stem cell fate.
METTL3 controls the differentiation fate of mesenchymal stem cells.
METTL3 is responsible for cancer stem cell functions.
Introduction
Stem cells are usually in an undifferentiated or not fully maturated state, which with capacities of continuously self-renewal and divide to produce offspring cells with specific functions, thereby with the potential to develop therapies for replacing defective or damaged cells resulting from a variety of disorders and injuries, such as Parkinson’s disease, heart disease, and diabetes1–4. Therefore, knowing the underlying mechanism for controlling stem cell capacities is beneficial for developing its application. Currently, understanding the mechanism of controlling the biological capacities of stem cells from the perspective of RNA N6-methyladenosine (m6A) modification has attracted great attention, which regulates gene functions post-transcriptionally and thereby affects cell functions5–8.
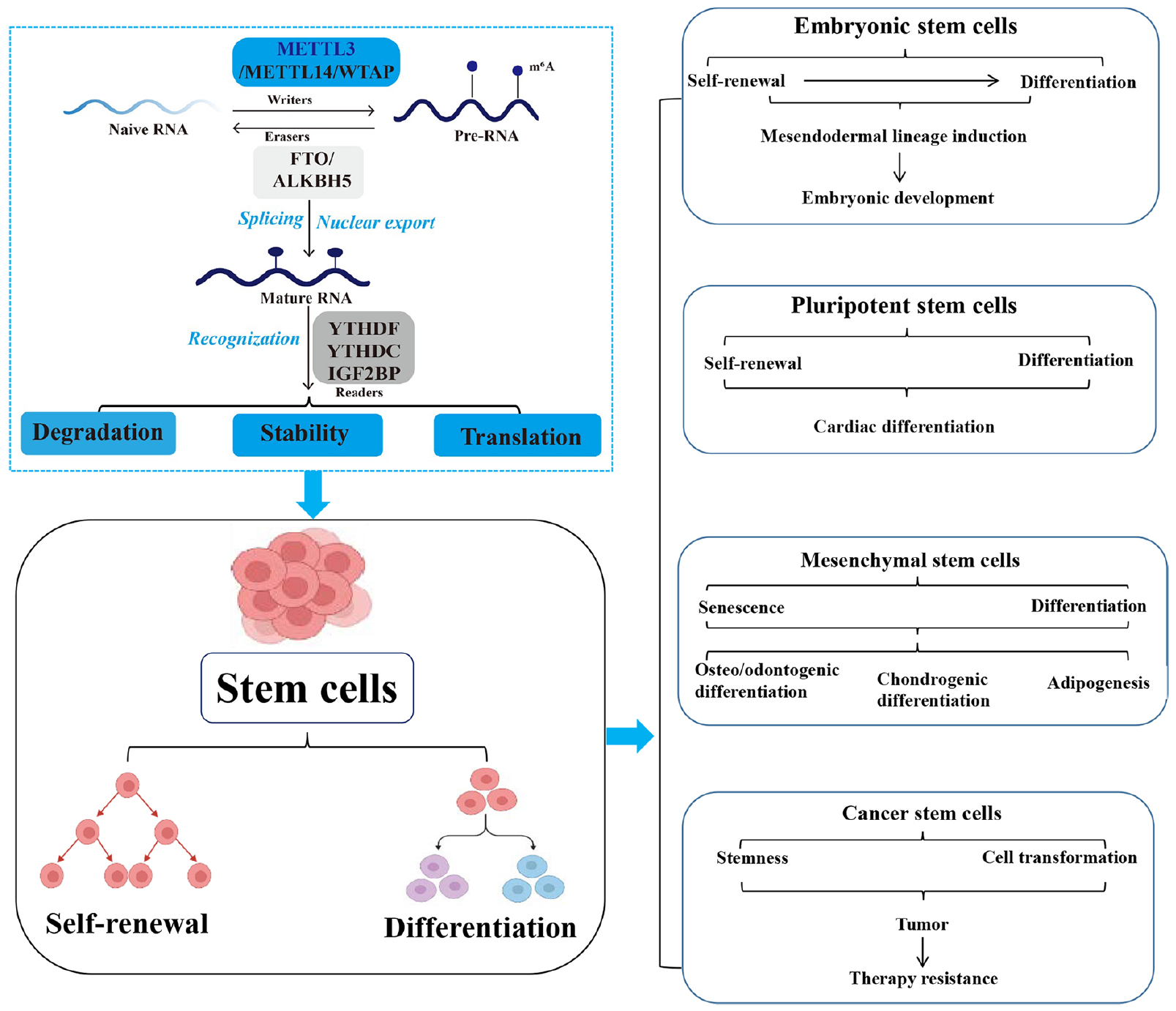
m6A, one post-transcriptional methylation modification capable of occurring on RNA adenine (A) of coding (mRNA) and non-coding [i.e., microRNA (miRNA), long non-coding RNA (lncRNA), circular RNA (circRNA)]9–11. The RNA m6A modification is a dynamic reversible process that functions depending on its component regulators called “writers” 12 , “erasers” 13 , and “readers” 14 . The writers, primarily constituted by the methyltransferase-like 3/14 (METTL3/14) and Wilms’ tumor 1-associating protein (WTAP), facilitate the N6-methylation of adenosine by employing S-adenosylmethionine as the methyl group donor 15 . In contrast, this methyl group is removed by erasers such as FTO (fat mass and obesity-associated protein) and ALKBH5 (AlkB homolog 5) 16 . The readers, typically such as YTHDF (YTH domain-containing family proteins), YTHDC (YTH domain-containing proteins), and IGF2BP (Insulin-like growth factor 2 mRNA binding protein), interact with the modified RNAs and then deciding the fate of RNA modifications, including splicing, nuclear export, stability, degradation, and translation, ultimately changing gene expression to regulate a variety of biological processes17–19.
Among these m6A regulators, METTL3, responsible for catalyzing m6A methylation of RNAs, plays important functional roles in contrast to the other regulators that are primarily essential for the m6A installation on RNAs20,21. METTL3-mediated m6A modification on RNAs has been extensively reported to regulate the biological functions of stem cells through different roles and mechanisms21,22. However, reviews that report the current status of the regulatory network of METTL3 in stem cells are lacking.
In this review, we summarize all available and original publications related to METTL3 and its role in the life cycle of stem cells. In addition, we aim to explain the status of METTL3 in regulating stem cells by systematically analyzing the functions and mechanisms of METTL3 within this context. This review might serve as a convenient reference for researchers to further investigate METTL3, as well as for developing the usages of METTL3 in stem cells (Fig. 1).
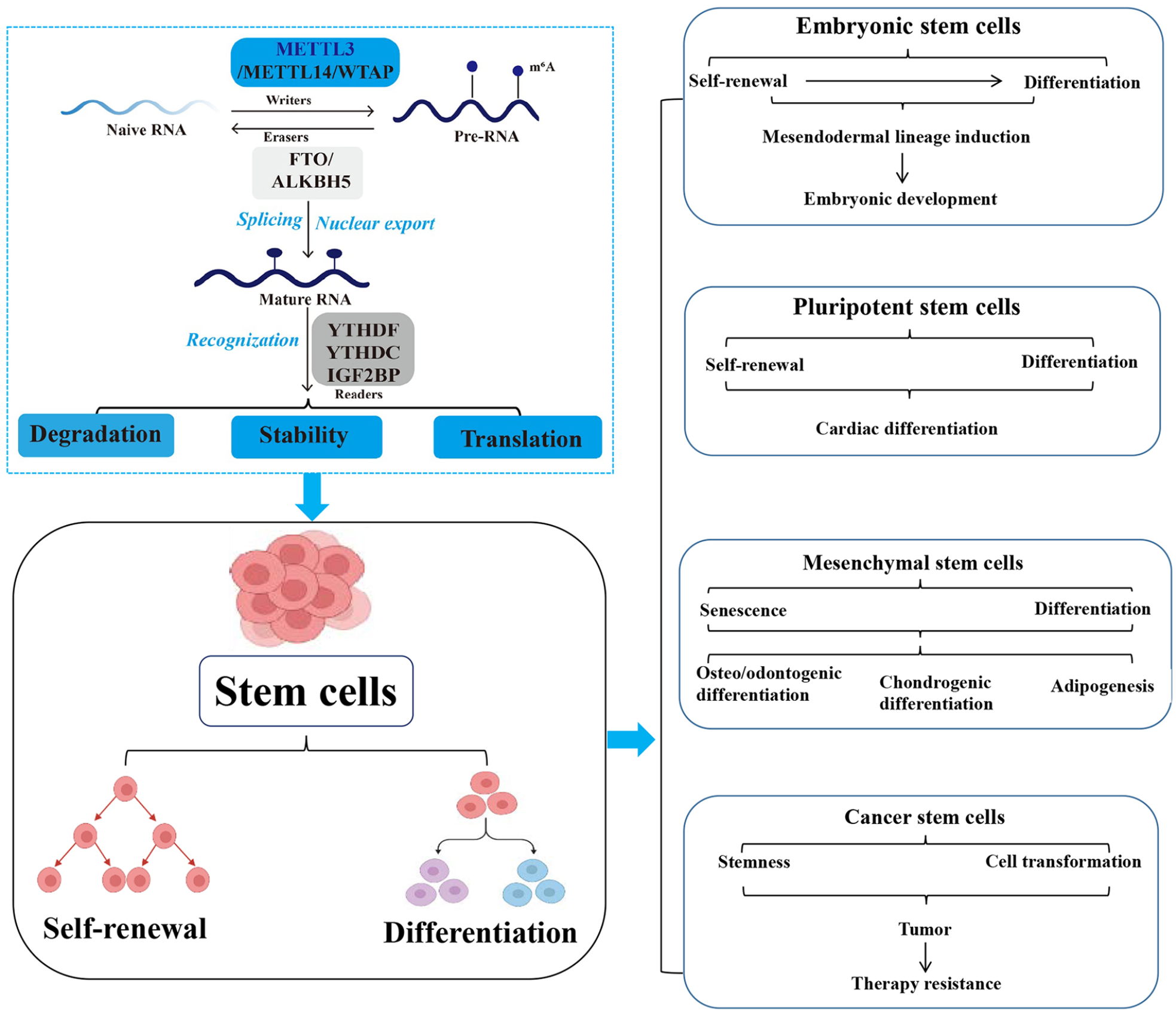
Graphical abstract. Roles and mechanisms of METTL3-mediated RNA m6A modification in stem cells, and their associated relationships and limitations. Highlights of clinical applications and future research directions for METTL3 in stem cells. The contribution of this review is to clarify the roles, mechanisms, and applications of METTL3 in stem cells. METTL3: methyltransferase-like 3; m6A: N6-methyladenosine; WTAP: Wilms’ tumor 1-associating protein; FTO: fat mass and obesity-associated protein; ALKBH5: AlkB homolog 5; YTHDF: YTH family proteins; YTHDC: YTH domain-containing proteins; IGF2BP: Insulin-like growth factor 2 mRNA binding protein.
Relationship Between METTL3 and Stem Cells
Generally, stem cells are primarily classified into embryonic stem cells (ESCs) and somatic stem cells according to their differentiation stage23–25. Furthermore, stem cells can be classified into pluripotent or unipotent based on their differentiation potential26–28. Although pluripotent stem cells (PSCs) can differentiate into any cell type, their differentiation has limitations 29 . Both ESCs and induced PSCs belong to PSCs, which with the ability of unlimited self-renewal and differentiation into almost any cell type of the body.
Stem cell renewal and differentiation are two intricately regulated processes during cell development and occur differently in stem cells according to their effects on cellular developmental processes 30 . Cell renewal primarily refers to mitosis, during which cells maintain their chromosome counts and produce daughter cells that are replicas of their progenitors 31 . Stem cell differentiation refers to the transformation of undifferentiated cells into specialized cells with specific functions 32 . Self-renewal generates pluripotent cells with regenerative tissue potential 33 . After differentiation, stem cells undergo metamorphosis into various specialized cell types 34 . These processes are regulated by genetic factors and signaling mechanisms. Therefore, any genetic alteration might result in an abnormal pattern of cellular renewal and differentiation, ultimately leading to abnormal cellular activity.
Currently, much attention has been devoted to the mechanisms of METTL3-mediated RNA m6A modification for explaining the stem cell transition between self-renewal and differentiation 35 . For instance, Chen et al. 36 demonstrated that the increased m6A abundance facilitates the reprogramming of mouse embryonic fibroblasts (MEFs) into PSCs. Moreover, manipulating the binding of METTL3 to the gene and cell type-specific mRNA transcripts further regulates the m6A levels 36 .
Functions and Mechanisms of METTL3 in ESCs
ESCs are stem cells that are derived from the inner cell mass of a mammalian embryo at a very early stage of development when it is composed of a hollow sphere of dividing cells (a blastocyst) 37 . ESCs from human embryos and embryos of certain other mammalian species can be grown in tissue culture 37 .
ESCs are particularly intriguing because of their ability to differentiate into a multitude of cell types, which is regulated by METTL3-mediated m6A modification on RNAs 38 . On the cusp of stem cell differentiation, m6A depletion by METTL3 knockout increases the likelihood of mesodermal lineage induction via activation of the p-Erk and phosphorylated Akt (p-Akt) signaling pathways 38 . Batista et al. 39 reported that the depletion of METTL3 in both mouse and human ESCs erases the m6A of Nanog mRNA, which leads to prolonged expression of Nanog during differentiation and impairs the ability of ESCs to exit the self-renewal stage toward the differentiation stage. Moreover, Aguilo et al. 40 reported that the chromatin-associated zinc finger protein 217 (Zfp217) interacts with METTL3 to promote the degradation of Nanog, Sox2, Klf4, and c-Myc mRNAs, which are linked to maintaining ESCs in an undifferentiated state. These findings suggest that METTL3-mediated m6A modification is a hallmark of transcriptome flexibility that is required for ESCs to differentiate into specific lineages. Additionally, Liu et al. 41 found that METTL3 is a crucial modulator for dynamic transcription and chromatin accessibility upon ESC-derived cardiac differentiation. Genome-wide analysis of chromatin-associated RNAs revealed that depletion of METTL3 enhances the transcription of pluripotent genes, as well as activates nascent cardiomyocyte-specific transcripts upon differentiation 41 . This impairs the ability of ESCs in self-renewal but promotes its differentiation. Furthermore, METTL3 negatively regulates the histone modifications on H3K4me3 and H3K36me3, which are involved in METTL3-modulated dynamic chromatin architecture during cell state transition 41 . Unexpectedly, nuclear m6A undergoes a dramatic increase upon differentiation, which correlates with the decrease in chromatin accessibility 41 . Collectively, these findings reveal that METTL3 and nuclear m6A epi-transcriptome couple with chromatin state to ensure transcriptional regulation of cell fate transition. Therefore, METTL3 is essential for maintaining ESC pluripotency.
m6A deposition on mRNA governs ESC fate by regulating the mRNA stability of pluripotency and lineage transcription factors 39 . Long-term single-cell tracking shows that Mettl3 knockdown in serum/leukemia inhibitory factor supports both pluripotency maintenance and its departure 42 . This is mediated by differential and opposing signaling pathways. Increased FGF5 mRNA stability activates p-Erk, leading to Nanog down-regulation. FGF5-mediated coactivation of p-Akt reinforces Nanog expression. In formative stem cells that are poised toward differentiation, m6A depletion by Mettl3 knockout activates both p-Erk and p-Akt, increasing the propensity for mesendodermal lineage induction 42 . Higher p-Erk counteracts the pluripotency exit delay that is exhibited by stably m6A-depleted cells upon differentiation. At single-cell resolution, the study illustrates that decreasing m6A abundance activates p-Erk and p-Akt-signaling, regulating pluripotency departure 42 . Furthermore, Sun et al. 43 found that Erk phosphorylates METTL3 at S43/S50/S525 and WTAP at S306/S341, followed by deubiquitination by USP5, resulting in the stabilization of the m6A methyltransferase complex. Lack of METTL3/WTAP phosphorylation reduces the decay of m6A-labeled pluripotent factors and traps mouse ESCs in the pluripotent state 43 . The same phosphorylation is also found in Erk-activated human cancer cells that contribute to tumorigenesis 43 . Overall, this study reveals an unrecognized function of Erk in regulating m6A methylation.
Furthermore, METTL3 regulates mouse ESC heterochromatin, the integrity of which is critical for silencing retroviral elements and for mammalian development44–46. METTL3 predominantly localizes to the intracisternal A particle (IAP)-type family of endogenous retroviruses. Knockout of Mettl3 impairs the deposition of multiple heterochromatin marks onto METTL3-targeted IAPs, and upregulates IAP transcription, suggesting that METTL3 is important for the integrity of IAP heterochromatin 44 . RNA transcripts derived from METTL3-bound IAPs are associated with chromatin and are m6A-methylated. These m6A-marked transcripts are bound by the m6A reader YTHDC1, which interacts with METTL3 and in turn, promotes the association between METTL3 and chromatin 44 . METTL3 also interacts physically with the histone 3 lysine 9 (H3K9) tri-methyltransferase SETDB1 and its cofactor TRIM28, and is important for their localization to IAPs. These findings demonstrate that METTL3-catalyzed m6A modification of RNA is important for the integrity of IAP heterochromatin in mouse ESCs, revealing a mechanism of heterochromatin regulation in mammals 44 . Additionally, Mettl3 is required for developmental pausing in mouse blastocysts and ESCs. Mettl3 enforces transcriptional dormancy by promoting global mRNA destabilization and suppressing the global nascent transcription by destabilizing the mRNA of the transcriptional amplifier and oncogene N-Myc, which is identified as a crucial anti-pausing factor 47 . Knockdown of N-Myc rescues the pausing in ESCs caused by Mettl3 deletion and forces demethylation and stabilization of Mycn mRNA in paused wild-type ESCs that largely recapitulates the transcriptional defects of Mettl3 deletion in ESCs 47 . These findings uncover Mettl3 as a key orchestrator of the crosstalk between transcriptomic and epi transcriptomic regulation during developmental pausing, with implications for dormancy in adult stem cells and cancer. Besides, depletion of METTL3 in blastocysts increases the mislocalization of heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2/B1) in ESCs by enhancing the m6A RNA methylation of the cell renewal markers OCT4 and SRY-box transcription factor 2 (SOX2), and by downregulating the expression of the cell differentiation marker GATA binding protein 4 (GATA4), ultimately results in developmental abnormalities 48 . Furthermore, Cao et al. 49 revealed that the knockdown of METTL3 in blastocysts causes m6A reduction, increasing the stability of autophagy-related 5 (ATG5) and LC3 mRNA, and elevating their expression solely in trophectoderm cells, impeding embryonic development during the morula-blastocyst transition, and leading to developmental abnormalities in trophectoderm cells 49 . These findings suggest that METTL3-mediated m6A methylation negatively affects autophagy within the trophectoderm lineage, which supports blastocyst development (Fig. 2).

METTL3 controls stem cell renewal and differentiation. Downregulation of METTL3 disrupts the normal transition between stem cell self-renewal and differentiation. Targets, mechanisms, and roles of METTL3 in stem cell renewal and differentiation are analyzed. METTL3: methyltransferase-like 3; p-Akt: phosphorylated Akt; IAP: intracisternal A particle; YTHDC: YTH domain-containing proteins; ZFP217: Zinc finger protein 217; SOX2: SRY-box transcription factor 2; ATG5: Autophagy-related 5; H3K9: Histone 3 lysine 9; GATA4: GATA binding protein 4; YTHDF: YTH family proteins; METTL14: methyltransferase-like 14; WTAP: Wilms’ tumor 1-associating protein.
Functions and Mechanisms of METTL3 in Pluripotent Stem Cells
In addition to regulating ESC differentiation and self-renewal, METTL3 also affects induced pluripotent stem cells (iPSCs). Wu et al. 50 demonstrated that METTL3 inhibition suppresses YTHDF1-mediated JAK2 translation and YTHDF2-dependent SOCS3 mRNA decay, resulting in the downregulation of JAK2 and upregulation of SOCS3, further inactivating the JAK2-STAT3 pathway, which weakens the self-renewal capacity and promotes iPSC differentiation. Furthermore, another study demonstrated that SMAD2/3 facilitates the recruitment of m6A MTC (METTL3/METTL14/WTAP) to specific transcripts that are involved in early human PSC fate determination, including the crucial pluripotency factor Nanog 51 . This activity primes specific transcripts for swift downregulation upon differentiation, ensuring a timely exit from pluripotency 51 . Specifically, the expression level of METTL3 is decreased during cardiomyocyte differentiation 38 . Deleting of Mettl3 at early stages of cardiac differentiation in mouse PSCs impedes m6A deposition, inactivating the expression of cardiac gene and postponing the outgrowth of embryoid bodies, whereas interfering m6A modification at later stages of differentiation shows minimal effects 38 . This study reveals a stage-specific requirement of METTL3-mediated m6A modification for the cardiac differentiation of PSCs and demonstrates that precise tuning of the m6A level is critical for cardiac differentiation (Fig. 2).
In summary, METTL3 is crucial for regulating the normal transition of PSCs between self-renewal and differentiation. Ultimately, this regulation facilitates the manipulation of embryonic formation, development, and degradation.
Functions and Mechanisms of METTL3 in Mesenchymal Stem Cells
Mesenchymal stem cells (MSCs) have been reported as effective for treating various diseases 52 . Wu et al. 53 found that both levels of m6A RNA methylation and METTL3 are reduced in prematurely senescent human MSC models of progeroid syndromes. Transcriptional profiling of m6A modifications further identifies MIS12, for which m6A modifications are reduced in both prematurely senescent MSCs and METTL3-deficient MSCs 53 . Knockout of METTL3 accelerates human MSC senescence, whereas overexpression of METTL3 rescues the senescent phenotype. Mechanistically, loss of m6A modification accelerates the turnover and decreases the expression of MIS12 mRNA; while knockout of MIS12 accelerates cellular senescence 53 . Furthermore, m6A reader IGF2BP2 is identified as a key player in recognizing and stabilizing m6A-modified MIS12 mRNA 53 . Taken together, METTL3 alleviates human MSC senescence through m6A modification-dependent stabilization of the MIS12 transcript, representing a novel epitranscriptional mechanism in premature stem cell senescence. MiRNAs play key roles in MSC differentiation 54 . Han et al. 55 discovered that METTL3 promotes osteo/odontogenic differentiation of stem cells by inhibiting miR-196b-5p maturation. Inhibition of miR-196b-5p promotes alkaline phosphatase (ALP) activity assay and mineralization in vitro, as well as enhances the expression of osteo/odontogenic differentiation markers DSPP and OCN in vivo 55 . Mechanistically, the results indicate that METTL3-dependent m6A methylation inhibits miR-196b-5p maturation by the microprocessor protein DGCR8. Moreover, miR-196b-5p negatively regulates METTL3 in stem cells of the apical papilla. Then, METTL3 strengthens the ALP activity assay, mineralization, and expressions of DSPP and OCN. Taken together, these findings highlight the critical roles of the METTL3-miR-196b-5p signaling axis in an m6A-dependent manner in osteo/odontogenic differentiation, identifying some potential targets for tooth and maxillofacial bone defects.
Wu et al. 56 found that METTL3-m6A methylase regulates the osteogenic potential of bone marrow mesenchymal stem cells (BMSCs) in osteoporotic rats via the Wnt signaling pathway. Lei et al. 57 found that METTL3 expression is upregulated in pro-inflammatory macrophages (M1), which promotes the secretion of IL-6 and iNOS from M1 macrophages. In the coculture condition, M1 macrophages with forced expression of METTL3 significantly enhance the migration ability of BMSCs, and also remarkably facilitate osteogenesis ability of BMSCs; the opposite is true when METTL3 is knockdown 57 . METTL3 silencing significantly reduces the m6A modification of HDAC5. Furthermore, HADC5 expression is downregulated in M1 macrophages with METTL3 knockdown 57 . In contrast, METTL3 overexpression promotes HDAC5 expression, indicating that HDAC5 is the target gene of METTL357. Therefore, METTL3 overexpression might be a new replacement therapy for bone repair. Besides, the silence of METTL3 decreases m6A methylation levels, inhibits osteogenic differentiation of BMSCs, and reduces bone mass 58 . METTL3-based m6A modification favors osteogenic differentiation of BMSCs through m6A-based direct and indirect regulation of RUNX2 58 . Furthermore, the chondrogenic differentiation of BMSCs has been used in treating and repairing cartilage defects. Yang et al. 59 found that Sox9, a critical transcription factor that mediates chondrogenic differentiation, exhibits enhanced translation by ribosome sequencing in chondrogenic pellets, which is accompanied by increased 5-methylcytosine (m5C) and m6A levels. Nsun4-mediated m5C and METTL3-mediated m6A modifications are required for Sox9-regulated chondrogenic differentiation 59 . Interestingly, in the 3′UTR of Sox9 mRNA, Nsun4 catalyzes the m5C modification and METTL3 catalyzes the m6A modification. Furthermore, Nsun4 and METTL3 co-regulate the translational reprogramming of Sox9 via the formation of a complex, which is assembled along with the recruitment of YTHDF2 and eEF1α-1. Moreover, BMSCs overexpressing METTL3 and Nsun4 promote the repair of cartilage defects in vivo. Taken together, this study demonstrates that m5C and m6A co-regulate the translation of Sox9 during the chondrogenic differentiation of BMSCs, which provides a therapeutic target in clinical settings. Adipogenesis of BMSCs promotes chemoresistance in acute myeloid leukemia (AML) cells. Pan et al. 60 found that METTL3 significantly inhibits BMSC adipogenesis. Mechanically, METTL3 affects AKT protein expression in BMSCs by mediating m6A modification on AKT1-mRNA. The downregulated expression of METTL3 in AML-BMSCs induces an increase in AKT expression, resulting in enhanced BMSC adipogenesis, thereby contributing to chemoresistance in AML cells 60 . Therefore, targeting AKT regulation by mRNA modification in BMSC adipogenesis might provide a novel therapeutic strategy to overcome AML chemoresistance.
Adipose-derived mesenchymal stem cells (ADMSCs), which are a type of adult MSC that exists in adipose tissue, have the potential for self-renewal and multidirectional differentiation 61 . Increasing research indicates that ADMSCs play a pivotal role in the process of wound repair 62 . METTL3 modulates osteogenic differentiation of ADSMCs in osteoporotic rats 63 . Zhou et al. 64 reported that ADMSCs accelerate the proliferation, migration, and lymphangiogenesis of lymphatic endothelial cells (LEC) via the METTL3 pathway and regulate VEGF-C expression via the METTL3/IGF2BP2-m6A pathway in diabetic foot ulcer (DFU) mice. VEGFR3 enhances ADMSC-mediated lymphangiogenesis via METTL3-mediated VEGF-C m6A modification to improve wound healing in DFU. Song et al. 65 found that METTL3 is upregulated and promotes ADMSC osteogenic differentiation. The m6A modification and expression of lncRNA RP11-44 N12.5 are both regulated by METTL3. Serine/threonine protein kinase 3 (STK3) is the predicted target gene of the lncRNARP11-44 N12.5. Subsequently, lncRNA RP11-44 N12.5 and METTL3 regulate the phosphorylation levels of three key proteins in the MAPK signaling pathway, ERK, JNK, and p38. In summary, METTL3 activates the MAPK signaling pathway by regulating the m6A modification and expression of lncRNA RP11-44 N12.5, thereby enhancing the osteogenic differentiation of human ADMSCs.
Functions and Mechanisms of METTL3 in Cancer Stem Cells
Stem cells can differentiate into malignant cells under unfavorable microenvironments 66 . Malignant tumors stem from the malignant transformation of stem cells or stem cell deterioration 67 . Cancer stem cells (CSCs) are associated with tumor growth, metastasis, and recurrence 68 . CSC accounts for a small subset of tumor cells with long-term tumorigenic capacity, which plays a pivotal role in cancer development and therapy resistance69,70.
METTL3 in CSC-induced cancer onset and progression
Increasing evidence shows that m6A modification contributes to the CSC phenotype by regulating gene expression 71 . CSC has been studied in multiple cancer types 72 . Moreover, multiple lines of evidence imply that METTL3 accelerates a variety of CSC phenotypes by regulating mRNA stability through m6A RNA methylation. For instance, Chen et al. 73 found that due to SOX4 transcriptional regulation, METTL3 regulates the malignant behavior of BMI1+ head and neck squamous cell carcinoma (HNSCC) stem cells through the cell division pathway. SOX4 regulates METTL3 expression in vitro. The progression and malignancy of HNSCC are decreased after Mettl3 is knocked out, while are increased after Mettl3 is knocked in Bmi1+ CSCs in vivo. Knockdown of Mettl3 inhibits stemness properties of CSCs in vitro. Mechanically, METTL3 mediated the m6A modification of ALDH1A3 and ALDH7A1 mRNA in Bmi1+ HNSCC CSCs. To sum up, regulated by SOX4, METTL3-mediated ALDH m6A methylation regulates the malignant behavior of BMI1+ HNSCC CSCs through the cell division pathway. Therefore, targeting METTL3 for CSC regulation might inhibit HNSCC tumor recurrence and metastasis. Consistently, Hexavalent chromium [Cr (VI)] is a common environmental carcinogen causing lung cancer in humans 74 . Wang et al. 75 found that chronic exposure to Cr induces the abnormal expression of METTL3, which in turn causes cell transformation, CSC-like property, and tumorigenesis. Similarly, both Arsenic and Benzo[a]pyrene (BaP) are Group I carcinogens. Exposure to either Arsenic or BaP can cause lung cancer 76 . Wang et al. 77 found that Arsenic plus BaP exposure alters RNA m6A methylation. Arsenic plus BaP exposure-transformed cells have significantly higher levels of RNA m6A methylation than Arsenic or BaP alone exposure-transformed human bronchial epithelial cells. Arsenic plus BaP exposure greatly upregulates METTL3 expression in cultured cells and mouse lung tissues. METTL3 knockdown in cells transformed by Arsenic plus BaP exposure drastically reduces their RNA m6A methylation levels, anchorage-dependent and -independent growth, CSC characters, and tumorigenesis. These findings suggest that Arsenic plus BaP co-exposure causes METTL3-mediated epi transcriptomic dysregulation, which contributes significantly to Arsenic plus BaP co-exposure-caused synergistic lung tumorigenic effect.
Glioma stem cells (GSCs) have high tumorigenic capacity performed by self-renewing ability78,79. METTL3 has been found to promote glioma development, maintain the self-renewal of GSCs, and participate in the regulation of drug resistance80,81. METTL3 functions as an oncogene in GBM by modulating nonsense-mediated (NMD) mRNA decay of splicing factors and alternative splicing isoform switches 82 . Lv et al. 83 found that PDGF ligands stimulate early growth response 1 (EGR1) transcription to induce METTL3 to promote GSC proliferation and self-renewal. Targeting the PDGF-METTL3 axis inhibits mitophagy by regulating m6A modification on optineurin (OPTN). The forced OPTN expression photocopies PDGF inhibition and OPTN levels portend longer survival of patients with GBM, suggesting a tumor suppressive role of OPTN. Pharmacologic targeting of METTL3 augments the anti-tumor efficacy of PDGF receptor (PDGFR) and mitophagy inhibitors in vitro and in vivo. Collectively, PDGF as upstream signaling of oncogenic m6A regulation drives tumor metabolism to promote CSC maintenance, highlighting PDGF-METTL3-OPTN signaling as a GBM therapeutic target. YY1 is highly expressed in GBM tissues and cells while silencing its expression reduces the self-renewal ability of GSCs. You et al. 84 found that YY1 promotes the transcriptional expression of SENP1 by binding to its promoter region, while the deSUMOase SENP1 facilitates the methylase activity of m6A through deSUMOylation of METTL3, thereby promoting the m6A modification of MYC mRNA and promoting MYC expression in vitro and in vivo. Conclusively, YY1 transcriptionally upregulates the SUMOylase SENP1 and enhances the methylase activity of METTL3, resulting in the increased m6A modification level of MYC mRNA, thereby promoting the self-renewal of GSCs.
Tumors that harbor an abundant CSC population may signal a poor clinical outcome in patients with hepatocellular carcinoma (HCC). Liver cancer stem cells (LCSCs) contribute to tumor recurrence and cancer cell proliferation in patients with HCC. Hu et al. 85 found that METTL3 is abnormally upregulated in LCSCs, which significantly correlates with individual cancer stage, tumor grade, and lymph node metastasis. Patients with low METTL3 expression have a longer overall survival time. Moreover, overexpression of METTL3 stimulates the proliferation and stemness of LCSCs in vitro and in vivo, while loss of METTL3 impedes it. Bioinformatics analysis determined that METTL3 modifies SOCS3 mRNA to reduce the stability of SOCS3 mRNA. Furthermore, METTL3 depletion suppresses the proliferation and stemness of LCSCs, in which SOCS3 overexpression is impaired and SOCS3 depletion facilitates the development of LCSCs via the JAK2/STAT3 pathway. Collectively, METTL3 facilitates the stemness and tumorigenicity of LCSCs by modifying m6A on SOCS3 mRNA. This study assumes that METTL3 facilitates LCSC proliferation and self-renewal by targeting SOCS3, which activates the JAK2/STAT3 signaling. CircMEG3 is downregulated and is negatively correlated with the expression of telomerase-related gene Cbf5 in human liver cancer, inhibiting telomerase activity and shortening telomere lifespan. Jiang et al. 86 indicated that CircMEG3 inhibits Cbf5 expression by METTL3 which depends on HULC. Strikingly, the increased Cbf5 abrogates CircMEG3’s ability to inhibit malignant differentiation of human LCSCs. Frizzled-10 (FZD10) is a 7-transmembrane-type WNT receptor, which is highly expressed in cancers. Wang et al. 87 found that METTL3-dependent m6A methylation of FZD10 mRNA activates FZD10 in LCSCs. FZD10 promotes self-renewal, tumorigenicity, and metastasis of LCSCs via activating b-catenin and YAP1. The FZD10-b-catenin/YAP1 axis is activated in LCSCs and predicts poor prognosis. Moreover, the FZD10-b-catenin/c-Jun axis transcriptionally activates METTL3 expression, forming a positive feedback loop (Fig. 3).
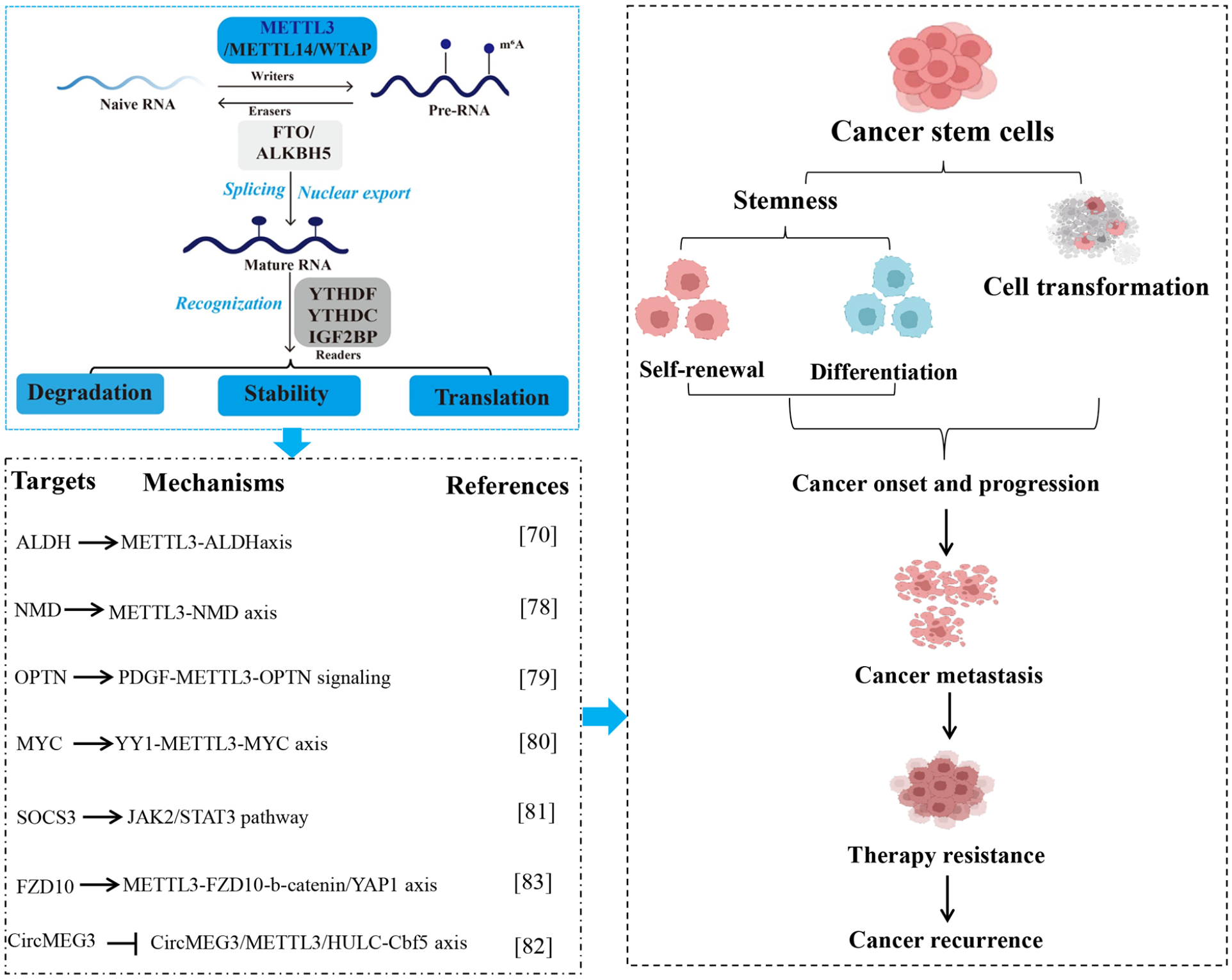
Functions and mechanisms of METTL3 in cancer stem cells. METTL3-mediated RNA m6A modification contributes to cancer occurrence, progression, metastasis, and treatment failure by regulating the self-renewal and differentiation of cancer stem cells. Targets, mechanisms, and roles of METTL3 in cancer stem cells are summarized. METTL3: methyltransferase-like 3; WTAP: Wilms’ tumor 1-associating protein; FTO: fat mass and obesity-associated protein; ALKBH5: AlkB homolog 5; YTHDF: YTH family proteins; YTHDC: YTH domain-containing proteins; IGF2BP: insulin-like growth factor 2 mRNA binding protein; NMD: nonsense-mediated; OPTN: optineurin; FZD10: frizzled-10.
METTL3 in CSC-induced cancer treatment failure
CSCs are important reasons for the failure of radiotherapy and chemotherapy 88 . Characterized by the ability to promote tumor growth and invasion, as well as tolerating radiation and chemotherapy, CSCs are self-renewing and insensitive to conventional therapies, leading to tumor recurrence 89 . For example, METTL3 accelerates Oxaliplatin resistance in CD133+ stem cells of gastric cancer by enhancing PARP1 mRNA stability 90 . Loss of METTL3 in CSC of bladder cancer restrained the angiogenesis and development of bladder cancer through m6A modulation on VEGF-A and TEK 91 .
GSCs have been considered as the “seeds” of GBM recurrence for their resistance to radiotherapy and chemotherapy78,79. Recent studies have consistently demonstrated that the high stemness and differentiation potential of GSCs leads to high heterogeneity, as well as tumor growth, immune escape, angiogenesis, and therapeutic resistance 92 . Temozolomide (TMZ) is the first-line and one of the most commonly used chemotherapeutic agents against GBM in clinical practice 93 . Chemo-resistance hinders the therapeutic efficacy of TMZ in treating GBM93. GBM-derived GSCs play vital roles in GBM resistance to TMZ. GSCs have the features of self-renewal, infinite proliferation, multi-potential differentiation, strong infiltration, and migration abilities 93 . GSCs also show high chemo-resistance. METTL3-mediated m6A modification is reported to be critical in GSC self-renew maintenance and radio-resistance. Shi et al. 94 found that the cell viability and half-maximal inhibitory concentration (IC50) of GSCs against TMZ are significantly decreased after GSC undergoes serum-induced differentiation to adherent growth of tumor cells. Besides, METTL3 expression and total m6A modification decline dramatically in consistency with GSC differentiation. Knockdown of METTL3 weakens self-renew, proliferation, and TMZ IC50 of GSCs, whereas enhancing TMZ-induced γH2AX level, indicating an upregulation of double-strand DNA damage. mRNA stability of two critical DNA repair genes (MGMT and APNG) is regulated by METTL3-mediated m6A modification 94 . In conclusion, METTL3-mediated m6A modification of MGMT and APNG mRNA plays crucial roles in suppressing TMZ sensitivity to GSCs, which suggests METTL3 is a potential new therapeutic target against GBM. Furthermore, Yin et al. 95 identified that LINC00839 is overexpressed in GSCs. A high level of LINC00839 is associated with GBM progression and radiation resistance. METTL3-mediated m6A modification on LINC00839 enhances its expression in a YTHDF2-dependent manner. LINC00839 functions as a scaffold to promote c-Src-mediated phosphorylation of β-catenin, thereby inducing Wnt/β-catenin activation. Combinational use of Celecoxib, an inhibitor of Wnt/β-catenin signaling, greatly sensitizes GSC to radiation.
Lenvatinib is approved as the first-line drug for advanced HCC. Analysis of patient cohorts, patient-derived tumor organoids, and patient-derived xenografts suggest that FZD10 might predict Lenvatinib’s clinical benefit in patients with HCC 87 . Furthermore, treatment of Lenvatinib-resistant HCC with an adeno-associated virus targeting FZD10 or a b-catenin inhibitor restores Lenvatinib response. Elevated FZD10 expression promotes the expansion of LCSCs and Lenvatinib resistance, which relate to METTL3 roles, indicating that FZD10 expression is a novel prognostic biomarker and MEETL3 is a therapeutic target for human HCC 87 .
CSC-related chemoresistance leads to poor outcomes for patients with colorectal cancer (CRC). Sec62 is originally found to be located in the membrane of the endoplasmic reticulum. Sec62 upregulation is associated with the chemoresistance of CRC and poor outcomes for patients with CRC. Depletion of Sec62 sensitizes CRC stem cells to chemotherapeutic drugs 96 . Mechanistically, the stabilization of Sec62 mRNA by METTL3-mediated m6A modification upregulates Sec62 expression, which subsequently promotes the stemness of CRC cells through competitively disrupting the interaction between β-catenin and APC to inhibit β-catenin degradation, thereby activating the Wnt/β-catenin signaling. In summary, METTL3-mediated m6A modification of Sec62 promotes the stemness and chemoresistance of CRC by binding to β-catenin and enhancing Wnt signaling. Thus, the METTL3-Sec62-β-catenin axis might act as a therapeutic target for improving CRC treatment.
Discussion
Stem cells intensively affect body development by their specific biological capacities such as self-renewal and differentiation. Understanding the mechanisms of stem cell properties will provide potential strategies for overcoming disease therapy, which requires finely controlled gene expression in various life cycle events 97 . METTL3, the catalytic subunit of the m6A RNA methyltransferase complex, orchestrates the expression and functionality of target genes through the modulation of various RNA processing mechanisms, including alternative splicing, nuclear export, translation efficiency, and RNA stability98,99. An increasing number of studies have reported the wide participation of METTL3-mediated modifications in stem cells. Here, we summarize these original publications and classify them by analyzing and interpreting the roles, mechanisms, and relationships of METTL3 during different events.
The analysis of the available publications suggests that METTL3 controls stem cell fates and is indispensable for maintaining its normal capacities. Specifically, METTL3 is required for regulating the transition between self-renewal and differentiation for several stem cells, including ESCs, PSCs, and MSCs. These processes ultimately contribute to the formation, development, and degradation of embryos. Therefore, METTL3 with the potential to control the function of stem cells in the translational and clinical fields.
Genetic alterations may cause abnormal cell renewal and differentiation patterns, which lead to cellular activity abnormalities. This review suggests that METTL3 is a key regulator for CSC stemness and cell transformation, which induces various cancers and contributes to cancer onset, progression, metastasis, therapy resistance, and recurrence. Therefore, METTL3 serves as the oncogene in CSC-induced cancers and with the potential for cancer diagnosis and prognosis in the clinic, as well as might be explored as a target for cancer treatment in future translational medicine.
To build on the established understanding of METTL3’s critical role in stem cell biology, it is essential to delve deeper into the mechanistic underpinnings of its function. This involves a meticulous examination of the molecular pathways and interactions that METTL3 engages in within different cellular contexts. One approach could be the use of advanced single-cell RNA sequencing technologies to capture the dynamic changes in gene expression profiles mediated by METTL3 in individual stem cells. This would provide a high-resolution map of METTL3’s impact on cellular heterogeneity, revealing potential subpopulations of stem cells that differentially respond to METTL3 activity. Moreover, the post-transcriptional modifications orchestrated by METTL3, such as m6A methylation, warrant further exploration to understand their precise roles in stem cell fate determination. For example, identifying the specific RNA targets of METTL3 and elucidating the downstream effects of their methylation status could uncover new regulatory nodes critical for stem cell maintenance and differentiation. Techniques such as crosslinking immunoprecipitation combined with high-throughput sequencing could be employed to map the binding sites of METTL3 on RNA transcripts, providing insights into the direct regulatory networks influenced by METTL3.
Additionally, the implications of METTL3 in tissue regeneration and repair are profound. Somatic stem cells possess remarkable potential for tissue regeneration and repair, yet their utilization in therapeutic contexts remains limited due to challenges in understanding their regulatory mechanisms. By manipulating METTL3 activity, it may be possible to enhance the regenerative potential of stem cells, offering new treatments for conditions such as neurodegenerative diseases, cardiovascular injuries, and musculoskeletal disorders. For instance, increasing METTL3 activity might promote the differentiation of iPSCs into functional neurons or cardiomyocytes, facilitating the repair of damaged tissues. Furthermore, the regulation of METTL3 activity could be pivotal in addressing age-related decline in stem cell function. As organisms age, the regenerative capacity of stem cells diminishes, contributing to tissue degeneration and the onset of age-related diseases. Understanding how METTL3 influences stem cell aging could lead to interventions that rejuvenate aging tissues and extend healthy lifespan. This could involve developing small molecules or gene editing techniques to modulate METTL3 activity specifically in aged stem cells.
Beyond its cellular functions, understanding METTL3’s interactions with other molecules can shed light on its wider physiological and pathological roles. It is crucial to consider METTL3’s interplay with the extracellular environment 100 . For example, studying how METTL3-driven m6A methylation impacts key signaling pathways and gene expression networks can uncover critical points that govern stem cell behavior and fate. This knowledge could advance our understanding of stem cell biology and improve precision medicine strategies. One promising research area is METTL3’s role in the tumor microenvironment. Since stem-like cancer cells drive tumor initiation, progression, and therapy resistance, understanding METTL3’s influence on these cells could revolutionize cancer treatment. Targeting METTL3 in cancer stem cells might inhibit their self-renewal and make tumors more responsive to conventional therapies, enhancing patient outcomes. Additionally, the extracellular matrix and signaling molecules in the stem cell niche significantly affect stem cell behavior. Exploring how METTL3-mediated modifications alter the secretion or reception of niche factors could provide new insights into the external regulation of stem cell fates. This research could utilize co-culture systems and three-dimensional organoid models to mimic complex in vivo conditions and evaluate METTL3’s impact on cell-cell and cell-matrix interactions.
Furthermore, the potential for METTL3 to serve as a biomarker in clinical settings should not be underestimated. Given its involvement in critical cellular processes, METTL3 expression levels and activity could provide valuable diagnostic and prognostic information for various diseases. For instance, assessing METTL3 expression in patient-derived samples could help stratify individuals based on their likelihood of responding to specific therapies, particularly in personalized medicine approaches for cancer treatment. Developing robust assays for METTL3 detection and quantification in clinical samples would be a crucial step toward its implementation as a biomarker. The therapeutic modulation of METTL3 presents both opportunities and challenges. While enhancing METTL3 activity might benefit regenerative medicine applications, inhibiting its function could be advantageous in oncology. However, achieving this duality requires the development of highly selective METTL3 modulators. Small molecule inhibitors or activators, designed through structure-based drug design and high-throughput screening, could offer precise control over METTL3 activity. Additionally, employing targeted delivery systems, such as nanoparticle-based carriers, could ensure that these modulators reach specific cell types or tissues, minimizing off-target effects and enhancing therapeutic efficacy.
In parallel, the potential side effects and safety concerns of targeting METTL3 should be thoroughly evaluated. Given its fundamental role in cell biology, indiscriminate modulation of METTL3 could have unintended consequences, such as affecting normal stem cell functions or inducing tumorigenesis. Therefore, future studies should aim to develop highly specific modulators of METTL3 that can precisely target pathological conditions without disturbing homeostatic processes. It is also imperative to consider the ethical and regulatory aspects of METTL3-targeted therapies. As with any novel therapeutic approach, rigorous preclinical and clinical testing must be conducted to assess the safety, efficacy, and long-term effects of METTL3 modulation. Ethical considerations regarding the manipulation of stem cell functions and the potential for unintended consequences must be addressed through transparent, multidisciplinary discussions involving scientists, clinicians, ethicists, and regulatory bodies. Finally, a comprehensive understanding of METTL3’s role across different species and tissue types will be crucial for translating these findings into clinical applications. Comparative studies in model organisms and human tissues can help identify conserved mechanisms and species-specific differences, guiding the design of effective and safe therapeutic strategies. Collaborative efforts integrating genomics, proteomics, and bioinformatics approaches will be essential in mapping the complex regulatory networks influenced by METTL3.
In summary, while the current understanding of METTL3’s role in stem cell biology and its therapeutic potential is promising, further research is required to fully realize its clinical applications. Detailed mechanistic studies, advanced technological approaches, and careful consideration of ethical and regulatory issues will be essential in harnessing METTL3’s potential. By continuing to explore and elucidate the complexities of METTL3-mediated regulation, we can unlock new avenues for innovative treatments that leverage the power of stem cells to address a wide range of human diseases and conditions.
Collectively, this review provides a systematic interpretation of how METTL3 functions in different stem cells and further clarifies their relationship. Therefore, this review establishes the regulatory network of METTL3 in stem cells, which provides a useful background as a reference for future exploitation of its clinical potential.
Footnotes
Author Contributions
YZ and FW contribute to the conceptualization and original draft preparation. YZ and SL contribute to reviewing and editing this manuscript. BS offers supervision and funding support.
Availability of Data and Materials
No datasets were generated or analyzed during the current study.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Sichuan Provincial Science & Technology Program (2022JDKP0040), Sichuan Provincial Health Commission Program (21PJ168), Deyang Municipal Science & Technology Program (2021SZZ068), and the College-level project of Chengdu University of Traditional Chinese Medicine (YYZX2021026 and YYZX2021020).