
Abstract
The role of mesenchymal stem cells (MSCs) in the breast tumor microenvironment (TME) is significant and multifaceted. MSCs are recruited to breast tumor sites through molecular signals released by tumor sites. Once in the TME, MSCs undergo polarization and interact with various cell populations, including immune cells, cancer-associated fibroblasts (CAFs), cancer stem cells (CSCs), and breast cancer cells. In most cases, MSCs play roles in breast cancer therapeutic resistance, but there is also evidence that indicates their abilities to sensitize cancer cells to chemotherapy and radiotherapy. MSCs possess inherent regenerative and homing properties, making them attractive candidates for cell-based therapies. Therefore, MSCs can be engineered to express therapeutic molecules or deliver anti-cancer agents directly to tumor sites. Unraveling the intricate relationship between MSCs and the breast TME has the potential to uncover novel therapeutic targets and advance our understanding of breast cancer biology.
Introduction
Breast cancer stands as the predominant malignant neoplasm among women, exhibiting a diverse spectrum of molecular characteristics and heterogeneity. According to the latest estimates provided by the American Cancer Society, it is anticipated that in the United States for the year 2023, a staggering number of approximately 297,790 new cases of invasive breast cancer will be diagnosed. In parallel, an estimated 55,720 new cases of ductal carcinoma in situ (DCIS) are expected to be identified. Tragically, the projected mortality rate from breast cancer stands at approximately 43,700 women 1 . These statistics underscore the criticality of addressing breast cancer as a significant public health concern and highlight the urgent need for effective prevention, early detection, and improved treatment strategies. The risk factors associated with breast cancer are complex, and we have categorized them into two distinct perspectives. The first category is lifestyle-related factors, which can be modified through self-discipline and conscious choices, including alcohol consumption, obesity, low levels of physical activity, no pregnancy or having the first child over 30 years old, insufficient breastfeeding, steroid hormones intake for birth control, or menopausal hormone therapy 2 . The second category encompasses inherent factors that are beyond individual influence. For example, older age, genetic mutations (BRCA1, BRCA2, CHEK2, PALB2, TP53, PTEN, CDH1, STK11, ATM, etc)3–5, family cancer history (particularly breast, ovarian, and other glandular cancers), personal benign or malignant breast lesions history, dense breast tissue, chest irradiation history, early menstrual periods and late menopause, and type II diabetes history 6 . Hence, it is imperative to minimize exposure to lifestyle-related factors on a population-wide scale and pinpoint women with elevated risk through precision prevention to mitigate the incidence of breast cancer.
Breast cancers exhibit heterogeneity, manifesting diverse histopathological and molecular features. Regarding histological types, there exist over 20 different classifications. The predominant one is invasive ductal carcinoma no special type (IDC-NST), while the second most prevalent is invasive lobular carcinoma (ILC) 6 . The grading process of breast cancer involves a microscopic evaluation of tubule formation, cellular pleomorphism, and mitotic index 7 . The widely used Nottingham grading system assigns a numerical score of 1–3 to each parameter and then calculates a total score to determine the grade 8 . Traditional TNM staging system incorporates clinical and pathological data related to tumor size (T), regional lymph node status (N), and the presence of distant metastases (M). Importantly, the newest staging system (AJCC-TNM8) integrates anatomic staging with tumor grade and the status of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) to determine a prognostic stage 9 . This prognostic staging system goes beyond merely assessing the physical extent of the disease and considers biological factors with predictive and prognostic significance. It offers more precise prognostic insights compared with previous staging systems 10 . The evaluation of ER, PR, and HER2 is a standard practice in breast cancer management because they are crucial predictive factors for hormonal and anti-HER2-targeted treatments 11 . Breast cancers that are ER/PR-positive (ER/PR+) tend to be of lower grade and less aggressive. While the majority of ER+ cancers also express PR, a small percentage of breast cancers exhibit positivity for only one of these hormone receptors, which appear to be more aggressive and less sensitive to hormonal therapy compared with those that are ER/PR+ 12 . Overexpression of HER2 is linked with a more aggressive clinical course and an unfavorable prognosis. However, it also serves as a predictive factor for a positive response to anti-HER2-targeted therapy 13 . Approximately, 15%–20% of breast cancers that lack expression of these three markers are categorized as triple-negative breast cancers (TNBCs). TNBCs typically display high-grade features, are associated with a less favorable prognosis, and exhibit limited responsiveness to targeted therapies 14 . Approximately, 15%–20% of TNBC cases are linked to BRCA1 or BRCA2 genes mutation 15 . Perou and colleagues introduced the first characterization of intrinsic molecular subtypes of breast cancer in 2000 through the application of cDNA microarray-based technology. They illustrated that, at the transcriptome level, breast cancer is not a uniform entity 16 . Five intrinsic subtypes are identified by global gene expression profiling studies, including luminal A, luminal B, HER2-overexpressing, basal-like and normal-like tumors 17 . Somatic mutations in TP53, PIK3CA, and GATA3 were observed in more than 10% of all breast cancer cases. The luminal A subtype exhibited the highest occurrence of significantly mutated genes, including PIK3CA (45%), followed by MAP3K1, GATA3, TP53, CDH1, and MAP2K4. In the case of luminal B, TP53 and PIK3CA mutations are the most prevalent (29% each). The HER2-overexpressing subtype is characterized by frequent HER2 amplification (80%), with high incidence of TP53 (72%) and PIK3CA mutations (39%). TP53 mutations occurred in 80% of basal-like cancers 18 . Researches dedicated to unraveling the molecular portraits of breast cancer continue to thrive. They are expected to drive progress and refinement in the molecular categorization of breast cancer, potentially resulting in alterations to treatment paradigms. The therapeutic strategies employed for breast cancer vary based on the phenotype, stage, grade, and molecular subtype, encompassing a range of locoregional interventions like surgery and radiation therapy, along with systemic treatment modalities, such as endocrine therapy for hormone receptor-positive tumors, targeted therapy, chemotherapy, and immunotherapy 19 .
MSCs are multipotent stem cells that have the potential to differentiate into several types of progenitor cells (such as adipocytes, osteoblasts, and myoblasts), and are involved in the maintenance and regeneration of connective tissues 20 . They can be separated from numerous tissue types, such as bone marrow (BM) 21 , adipose tissue (AT) 22 , dental pulp 23 , peripheral blood 24 , hair follicle 25 , lungs 26 , the placenta 27 , the umbilical cord (UC) 28 , UC blood, Wharton’s jelly 29 , amnion 30 , and chorion 31 . Among these, MSCs derived from BM, AT, and UC are the three most utilized in preclinical and clinical research 32 . The biological traits of MSC determine its application in clinical. First, MSCs can migrate toward and engraft in inflammation sites. Then MSCs interact with surrounding cells by paracrine signaling or direct physical contact. Moreover, MSCs have the potential to differentiate into epithelial cells 33 , osteoblast 34 , cardiomyocytes 35 , neuron cells 36 , islet cells 37 , hepatocytes 38 , and so on. MSCs demonstrate broad clinical applicability, spanning diverse fields, including cancers, heart disease, liver disease, lung disease, diabetes, Crohn’s disease, brain disease, spinal cord injury, graft versus host disease, and more 39 . We will focus on the therapeutic application of MSCs in breast cancer.
The progression of cancer is influenced not only by the inherent qualities of the cancer cells themselves but also by the composition of the tumor microenvironment (TME) and the interaction between its various cellular components and the cancer cells 40 . As a result, the complexity of the breast cancer microenvironment plays a significant role in determining the outcome for the patient 41 . The neoplasm proliferates within a permissive microenvironment where diverse biomolecules, including cytokines, chemokines, and growth factors, are strategically positioned. In addition, a variety of cellular components, such as breast cancer stem cells (BCSCs), tumor-associated macrophages, MSCs, cancer-associated fibroblasts (CAFs), lymphocytes, dendritic cells (DCs), and neutrophils, collaboratively contribute to the establishment of optimal conditions favorable for cancer development42,43. The orchestrated interplay among these biomolecules and cells functions to promote tumor growth, angiogenesis, epithelial–mesenchymal transition (EMT), and metastasis44,45. In particular, the adjacent AT offers a plentiful supply of MSCs that play a significant role in the stromal components of the breast TME 46 . In addition, given the multiple similarities between the development of cancer and wound healing, tumors are often referred to as “wounds that do not heal” 47 . Like sites of damage, breast tumor tissues attract MSCs by releasing different endocrine and paracrine signals 48 . Meanwhile, multiple experiments have demonstrated that MSCs interact with breast cancer cells. For example, when breast cancer cells were treated with a medium containing MSCs-derived extracellular vesicles (EVs), the expression of E-cadherin was reduced and the level of N-cadherin was increased, which promoted the EMT 49 . Therefore, it is essential to understand how specific components within the TME interact, how cells communicate within this network, and how cancer cells interact with other cell populations in the TME.
In this context, our focus is on the biological behavior of MSCs in breast cancer, including their migration into the TME, polarization within the TME, communication within the cellular network, and potential applications in breast cancer treatment.
Tumor Homing Ability of MSCs
Homing is a biological process in which cells migrate toward specific tissues in the body 50 . MSC homing refers to the migration of MSCs toward sites of injury or inflammation, serving as a physiological mechanism to maintain tissue homeostasis and facilitate tissue regeneration51,52. It has been discovered that MSCs spontaneously distribute in various nonhematopoietic tissues after systemic infusion in baboons, indicating their selective migration ability to specific organs 53 . A study in mice found that MSCs migrated toward the tumor sites regardless of how they were injected 54 . The administration of MSCs into the bloodstream of mice bearing MCF7 or MDA-MB-231 cells significantly attracted MSCs to the tumor sites, with limited migration to critical organs, such as the liver, kidneys, and spleen 48 . However, the ability of these breast cancer cell subtypes to attract MSCs varied. For example, the highly invasive MDA-MB-231 cells were found to be more effective in promoting migration of MSCs in vitro and in vivo compared with the weakly invasive MCF-7 cells 55 . Current knowledge suggests that functional molecules, including cytokines, chemokines, and growth factors work together to facilitate the tumor homing ability of MSCs (Fig. 1).

Homing characteristics of MSCs. MSCs, commonly derived from bone marrow, adipose tissue, and dental pulp, show a remarkable and specific affinity for tumor sites. Mechanically, soluble factors secreted by tumor cells, such as TNF-α and IFN-γ, drive MSCs to adhere to vascular endothelium by upregulating VCAM-1 and P-selectin. Simultaneously, in response to cytokines and growth factors from tumor cells, including IL-6, IL-8, PDGF-BB, VEGF, and so on, MSCs enhance the expression of CXCR4, CXCL7, and receptors for corresponding growth factors. As a result, MSCs aggregate in breast tumor microenvironments and exert an effect on tumor biology. MSC, mesenchymal stem cells; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; VCAM-1, vascular cell adhesion molecule-1; IL-1β, interleukin-1β; IL-6, interleukin-6; IL-8, interleukin-8; PDGF-BB, platelet-derived growth factor-BB; VEGF, vascular endothelial growth factor; CXCR4, C–X–C chemokine receptor type 4; CXCL7, C–X–C motif chemokine ligand 7; PDGFR-β, platelet-derived growth factor receptor-β; VEGFR, vascular endothelial growth factor receptor; FGFR, fibroblast growth factor receptor; CCL2, C–C motif chemokine ligand 2; FGF2, fibroblast growth factor 2; HDGF, hepatoma-derived growth factor.
The initiation of MSC homing to the tumor site involves two crucial steps: the adhesion of MSCs to the vascular endothelium, followed by their passage through the endothelial layer. Cytokines are believed to play a role in facilitating this process. Specifically, tumor necrosis factor-α (TNF-α) is known to upregulate the expression of vascular cell adhesion molecule-1 (VCAM-1) on MSCs, which, in turn, promotes their adhesion to endothelial cells (ECs) 56 . In addition to TNF-α, the interaction between MSCs and ECs is facilitated by molecules like P-selectin and VCAM-1. This interaction is essential for the extravasation of MSCs into tumor tissue 57 . Other inflammatory cytokines, such as interleukin (IL)-1β and interferon-γ (IFN-γ), can also induce the expression of VCAM-1, thereby contributing to the accumulation of MSCs 58 . In breast cancer, the secretion of IL-6 by SUM149 and MCF-7 cells leads to the binding of IL-6 to IL6R/GP130 receptors on MSCs, resulting in MSCs producing CXCL7 and undergoing chemotaxis. CXCL7, in turn, induces the secretion of multiple cytokines, such as IL6, IL8, CXCL6, and CXCL5, forming a positive feedback loop 59 . In the hypoxic conditions of the breast TME, tumor cells increase IL-6 secretion, further promoting MSC migration through the activation of STAT3 and MAPK signaling pathways 60 . In addition, the activation of CXCR4/SDF-1 axis leads to MSCs migration to the tumor sites in the 4T1 mouse model 61 . MDA-MB 231 and MCF-7 cells exhibit a substantial upregulation of IL-8 expression. When exposed to IL-8, breast resident AT-derived MSCs become more invasive toward tumor-conditioned medium. The recruitment of MSCs may be related to the negative regulation of RECK signals, which can subsequently upregulate the expression of matrix metalloproteinase (MMP) and increase cell migration 62 .
In addition to inflammatory factors, chemokines also facilitate the migration of MSCs to tumor tissue. Monocyte chemotactic protein-1 (MCP-1 or CCL2) is a chemokine consisting 76 amino acids. Dwyer et al. investigated substances secreted by primary breast tumors and found that MCP-1 mediates the homing of MSCs to the site of primary breast cancer. Notably, Claudin low-type MDA-MB-231 cells express more MCP-1/CCL2 than luminal A-type T47D cells 63 , potentially explaining the higher ability of aggressive breast cancer cells to attract MSCs for migration 64 . Furthermore, studies have shown that exposure to radiation can increase the production of MCP-1/CCL2, consequently enhancing tumors’ ability to attract MSCs 65 . This finding further underscores the significant role of MCP-1/CCL2 in recruiting MSCs toward tumors.
Third, growth factors play a significant role in this context. MDA-MB-231 cells and 4T1 cells produce platelet-derived growth factor-BB (PDGF-BB), which promotes the migration of MSCs by binding to platelet-derived growth factor receptor-β (PDGFR-β) on MSCs. The process can be blocked by neutralizing antibody against PDGFR-β 66 . In addition, two other growth factors, vascular endothelial growth factor (VEGF) and fibroblast growth factor 2 (FGF2), have relevance in MSC migration toward breast cancer. They are produced by tumor cells. MSCs are found to express their receptors. Depletion of VEGF and FGF2 has been shown to reduce MSC migration 55 . Hepatoma-derived growth factor (HDGF), a protein with heparin-binding affinity, has been found to be upregulated in MDA-MB-231 cells and can facilitate MSC chemotaxis 67 . Furthermore, cyclophilin B exhibits similar biological activity toward MSCs 67 .
Finally, it is important to note that the ability of MSCs to migrate toward tumors is not exclusively influenced by signals from breast cancer cells. A study has shown that treatment with histone deacetylase inhibitors can increase the expression of urokinase plasminogen activator (uPA) in UC-derived and BM-derived MSCs. This elevated uPA expression enhanced the ability of these MSCs to be attracted by prostate and breast tumor cells when compared with their vector-treated counterparts, suggesting that the inherent features of MSCs may impact their efficiency in being recruited to tumors 68 .
Polarization of MSCs and Its Dual Role in Breast Cancer Progression
Although progress has been made in understanding how MSCs function within the TME, there are still many questions about whether MSCs are safe and effective for clinical use. This is due to the two-sided nature of their signals once they are recruited to the different tumor models and microenvironment. For example, in human ovarian cancer, tumor-associated MSCs (T-MSCs) were discovered to produce more bone morphogenetic proteins (BMPs) compared with normal MSCs (N-MSCs) derived from AT and BM, including BMP2, BMP4, and BMP6 69 . Studies indicated that BMP2/4 can promote the progression of various types of cancers, such as prostate 70 , melanoma 71 , and breast cancer 72 . Koushan et al. isolated MSCs from stage II breast tumor samples that had undergone chemotherapy or radiotherapy. They found that T-MSCs and N-MSCs exhibited similar appearance and immunophenotype; however, they displayed distinct differences in certain immunomodulatory functions and protein production. Compared with N-MSCs, T-MSCs secreted higher levels of transforming growth factor beta (TGF-β), prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase (IDO), and VEGF, while showing reduced levels of MMP-2 and MMP-9 73 , 74 . T-MSCs also promoted peripheral blood lymphocytes (PBLs) to secrete immunosuppressive factors, including IL-10, TGF-β, and PGE2 75 . Conversely, T-MSCs enhanced the proliferation of PBLs and had no impact on the polarization of regulatory T cells (Tregs) 75 . The conflicting results may help to explain their dual roles in both tumorigenesis and immune modulation.
The mechanisms of MSCs polarization remain unclear. Interestingly, it is suggested that T-MSCs may have the ability to transform newly arrived naive BM-MSCs into T-MSCs, resulting in the expansion of T-MSCs within the TME. Co-culturing BM-MSCs with T-MSCs causing BM-MSCs to acquire a similar tumor-promoting capability to that of T-MSCs 76 . Furthermore, a study conducted by Ren et al. suggested the role of TNF-α in polarizing normal tissue MSCs into the tumor-promoting phenotype. They observed that when BM-MSCs were pretreated with TNF-α, BM-MSCs produced similar chemokines to T-MSCs and promoted tumor growth in EL-4 lymphoma, B16 melanoma, and 4T1 breast carcinoma models 77 . It has also been demonstrated that BM-MSCs acquire the tumor-promoting characteristics after exposure to MDA-MB-231 breast tumor-conditioned medium for 30 days, leading to an upregulated expression of stromal-derived factor-1 (SDF-1) 78 . In addition, activation of TLR3 and TLR4 (Toll-like receptors) can influence the properties of MSCs. TLR3 stimulation tends to transform MSCs into an immunosuppressive phenotype, while TLR4 stimulation induces a proinflammatory phenotype 79 . CD90 could potentially serve as a novel indicator of MSCs status. It was observed that CD90low AT-MSCs inhibited E0771 tumor growth compared with CD90high AT-MSCs 80 .
Once MSCs migrate to the TME and differentiate into distinct phenotypes, they can exert both positive and negative effects on breast cancer progression (Fig. 2). When AT-MSCs were directly injected into a mouse model of MDA-MB-231 cells, it resulted in increased tumor volume, elevated accumulation of Ki67, and reduced Beclin expression within the tumor tissues 81 . In in vivo studies involving the injection of mixed cell populations comprising breast cancer cells and BM-MSCs into mice, it was found that CCL5, derived from MSCs, primarily acts on MDA-MB-231 cells in a paracrine manner, promoting tumor cell metastasis via CCL5-CCR5 signaling pathway. In addition, MSCs enhance the growth of MCF7/Ras tumor 48 . AT-MSCs have been shown to promote the proliferation of MDA-MB 231 cells and activate the mTOR expression. They also enhance the growth of 4T1 tumors 81 . Collagen receptor, discoidin domain receptor 2 (DDR2) derived from MSCs results in an upregulation of DDR2 in breast cancer cells, ultimately promoting metastasis and growth. In mice lacking the ddr2 gene and carrying slie mutation, tumors do not spread to other parts 82 . AT-MSCs trigger the process of EMT in MCF7 cells through the production TGF-β1 83 . Furthermore, the activation of ERK pathway in MDA‑MB‑231 and MCF‑7 cells, induced by umbilical cord-derived MSCs (UC-MSCs), is also associated with the augmentation of EMT 49 .
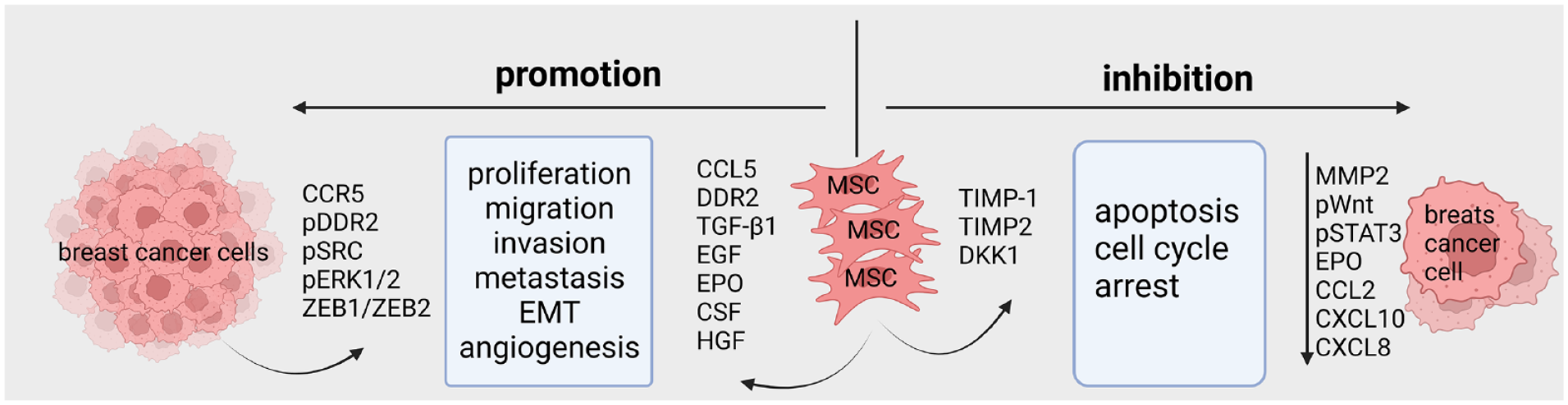
MSCs exert both positive and negative effects on breast cancer progression. After migrating to the tumor site, MSCs undergo polarization into T-MSCs, demonstrating the capability to secrete a spectrum of pro-tumorigenic factors, including CCL5, TGF-β, EGF, HGF, and so on. This, in turn, leads to the phosphorylation of DDR2, Src, and ERK1/2 in breast cancer cells, thereby promoting tumor proliferation, migration, invasion, angiogenesis, and EMT. Nevertheless, certain findings have also demonstrated that MSCs induce apoptosis and enforce cell cycle arrest in breast cancer cells by downregulating MMP-2 and the phosphorylation levels of Wnt and STAT3 in cancer cells, consequently impeding neoplastic progression. EMT, epithelial–mesenchymal transition; MSCs, mesenchymal stem cells; T-MSCs, tumor-associated MSCs; CCL5, chemokine (C–C motif) ligand 5; TGF-β, transforming growth factor beta; EGF, epidermal growth factor; HGF, hepatocyte growth factor; DDR2, discoidin domain receptor 2; MMP-2, matrix metalloproteinase 2; CCR5, C–C chemokine receptor type 5; EPO, erythropoietin; CSF, colony-stimulating factor; TIMP, tissue inhibitor of metalloproteinase; DKK1, dickkopf 1; CCL, C–C motif chemokine ligand; CXCL, C–X–C motif chemokine ligand.
However, there is growing evidence suggesting that MSCs could potentially inhibit the progression of breast cancer. It has been shown that MSCs produce proteins, TIMP-1 and TIMP-2 (tissue inhibitor of metalloproteinase), which possess anti-metastatic properties 84 . Notably, Clarke et al. proposed a potential explanation for the dual effects that MSCs can exert on tumor growth. Initially, MSCs secrete MMP-2, which has a pro-migratory and pro-metastatic impact on tumor cells. However, over time, the increased release of TIMP-1 and TIMP-2 inhibit the activity of MMP-2, thereby shifting the balance toward an anti-invasive effect 84 . Various models have demonstrated the tumor-suppressive effects of MSCs, including co-injection of MSCs and MDA-MB-231 cells, as well as intravenous injection of MSCs after cancer cells inoculation. Notably, UCB-MSCs demonstrate greater efficacy in inhibiting tumor progression to AT-MSCs 85 . Furthermore, research has shown that UCB-MSCs can inhibit the growth of MDA-MB-231 cells by upregulating dickkopf 1 (DKK1), thus inhibiting the Wnt signaling pathway. In addition, MSC-derived extracellular matrix (ECM) is effective in halting tumor growth by increasing the level of PTEN in cancer and activating AKT signaling pathway 86 . To provide a comprehensive overview, we have summarized the contradictory roles of MSCs isolated from various tissues, their markers, and their effects on breast cancer cells in Table 1.
The Dual Roles of MSCs in Breast Cancer.

MSC, mesenchymal stem cells; BM, bone marrow; CCL, C–C motif chemokine ligand; AT, adipose tissue; DDR2, discoidin domain receptor 2; DP, dental pulp; EMT, epithelial–mesenchymal transition; TIMP, tissue inhibitor of metalloproteinase; UC, umbilical cord; UCB, umbilical cord blood; EGF, epidermal growth factor; EPO, erythropoietin; CSF, colony stimulating factor; HGF, hepatocyte growth factor; PDGF-AA, platelet-derived growth factor-AA; CXCL, C-X-C motif chemokine ligand.
The Cellular Network of MSCs in TME
After MSCs homing and polarization in breast tumor sites, T-MSCs produced higher levels of TGF-β, IDO, and VEGF, which enable them to adapt and perform different roles depending on the specific conditions of TME 75 . Here, we summarize the interactions of MSCs with various cells present in breast TME, including immune cells, CAFs, cancer stem cells (CSCs), and cancer cells (Fig. 3).

The cellular network of MSCs in breast tumor microenvironment. The progression of breast cancer is influenced not only by the inherent qualities of the cancer cells themselves, but also by the composition of the TME and the interaction between its various cellular components and the cancer cells. MSCs are recruited to breast tumor sites through molecular signals released by tumor sites. Once in the tumor microenvironment, MSCs undergo polarization and interact with various cell populations, including immune cells, CAFs, BCSCs, and breast cancer cells. MSCs exhibit the ability to modulate immune responses and create an immunosuppressive environment, thereby influencing the tumor immune landscape. In addition, their interaction with CAFs and BCSCs contributes to the promotion of tumor progression and self-renewal capabilities. Furthermore, MSCs establish communication networks with cancer cells, affecting tumor cell behavior and characteristics, such as proliferation, migration, EMT, and dormancy. The complex interplay between MSCs and the components of the breast tumor microenvironment highlights the need for further investigation. MSC, mesenchymal stem cell; TME, tumor microenvironment; CAF, cancer-associated fibroblast; BCSC, breast cancer stem cell; EMT, epithelial–mesenchymal transition; DC, dendritic cell; BCC, breast cancer cell; TGF-β, transforming growth factor beta; HGF, hepatocyte growth factor; PDL1/2, programmed death-ligand 1/2; IL, interleukin; CCL5, chemokine (c–c motif) ligand 5; TNF-α, tumor necrosis factor-α; ALDH1, aldehyde dehydrogenase 1; CXCL7, chemokine (c–x–c motif) ligand 7; SDF-1, stromal-derived factor-1; CXCR4, c–x–c chemokine receptor type 4; VEGF, vascular endothelial growth factor; DDR2, discoidin domain receptor 2; BMP7, bone morphogenetic protein 7.
Interactions Between MSCs and Immune Cells
A properly functioning innate immune system is essential to prevent abnormal cell populations from growing uncontrollably in an environment that promotes cancer progression. Immune cells, such as DCs, macrophages, and neutrophils play a key role in activating adaptive immune response and elimination of cancer cells. The differentiation of BM progenitors into DCs when cultured with conditioned supernatants from MSCs was partially hindered due to the secretion of IL-6 by MSCs. This resulted in a reduced expression of MHCII, CD40, and CD86 co-stimulatory molecules on mature DCs, leading to decreased T cell proliferation 91 . Mechanically, IL-10 generated by T-MSC suppresses the expression of cystathionase gene in DCs through the IL-10/STAT3 signal, resulting in the inhibition of T cell proliferation and IFN-γ secretion 92 . Furthermore, genetically engineered MSCs that express TNF-α/CD40L were found to promote the maturation of DCs when they were co-cultured together 93 . In addition, the co-injection of DCs and engineered MSCs effectively inhibited the growth of 4T1 cells and prolonged survival in a BALB/c mouse model 93 .
In addition to DCs, MSCs also influence the biological characteristics of macrophages. Exosomes originated from cancer cells empower MSCs to enhance the infiltration of macrophage into B16-F0 melanoma or EL-4 lymphoma
94
. Upon arrival, these macrophages polarize toward a pro-tumor M2-like phenotype under the influence of molecules, such as CCL2, CCL7, and CCL12, which are produced by T-MSCs
77
. In Brpkp110 breast tumors, the conversion of macrophages into the M2 phenotype is associated with factors derived from MSCs, including TGF-β, C1q, and semaphorins. This leads to an upregulation in the expression of
Neutrophils play a crucial role as the initial defense mechanism against infections and tissue injury. In infectious disease, placenta-derived MSCs (P-MSCs) significantly enhance the phagocytic capacity of neutrophils by releasing IL-1β. When P-MSCs are administered to Hypervirulent Klebsiella pneumoniae-infected mice, they effectively attenuate T and natural killer (NK) cell responses. Intriguingly, P-MSCs exhibit a selective capacity to recruit neutrophils and promote their antibacterial functions, ultimately contributing to the survival of the mice 96 . However, in the context of cancer, the secretion of inflammatory factors, such as IL-17, IL-23, and TNF-α by tumor-associated neutrophils activate the AKT and p38 pathways in MSCs, leading to the transformation of MSCs into CAFs. This transformation significantly promotes the growth and metastasis of gastric cancer 97 . When neutrophils pretreated with MSCs were injected into the 4T1 cancer model, it resulted in accelerated tumor growth. This was attributed to MSCs enhancing the arginase activity of neutrophils, which subsequently exhibited an immunosuppressive phenotype, leading to the inhibition of T cell proliferation 98 .
In addition to their indirect interactions with immune cells, as mentioned earlier, MSCs have been discovered to exert a direct influence on the functions of T cells. MSCs can modulate T cell responses by secreting various soluble factors that possess immunosuppressive properties. Notably, MSC-secreted factors, such as TGF-β and HGF, have been implicated in inhibiting the proliferation of both T cells and B cells 99 . Through the secretion of TGF-β1 and Th2-type cytokines, MSCs enhance the infiltration of Tregs in cultures containing breast cancer T47D cells and lymphocytes, consequently inhibiting the function of NK cells and cytotoxic T cells (CTLs) 100 . Furthermore, MSCs impede the proliferation of CD4+ T cells and CD8+ T cells by reducing the expression of surface markers, including CD25, CD38, and CD69, which serve as indicators of T cell activation 101 . Zhang et al. demonstrated that MSCs significantly impaired the CAR-T cell-mediated cytotoxicity through the secretion of staniocalcin-1, leading to an increase in CD4+ T cells and Treg cells while reducing the population of CD8+ T cells. Moreover, MSCs induced the expression of immunosuppressive factors, such as IDO and PD-L1, thereby contributing to the establishment of an immune-suppressive TME 102 . MSCs also inhibited the activation of CD4+ T cells by direct cell contact and the production of PD-L1 and PD-L2. This, in turn, led to a decrease in the secretion of IL-2 and induced T cells apoptosis 103 .
Interactions Between MSCs and CAFs
CAFs are abundant in the breast tumor stroma, and research has demonstrated their significance in breast cancer progression 104 . Notably, there are distinct phenotypic differences observed in CAFs among various molecular subtypes of breast cancer. The gene expression profiles of CAFs in HER2-positive breast cancer, in contrast to TNBCs and ER+ breast cancer, exhibit substantial variations. Specifically, genes linked with cell skeleton and integrin signaling were found to be significantly upregulated in HER2-positive breast cancer, correlating with increased tumor migration and poorer prognosis 105 . In cases of ILC and TNBC, CAFs are shown to originate from normal fibroblasts (NFs) within breast tissue. CD26-positive NFs undergo a transformation into inflammatory CAFs (iCAFs), which is a pro-tumor phenotype. iCAFs promote breast cancer progression by attracting myeloid cells through the production of CCL2 and by enhancing invasive activity via MMPs 106 . Furthermore, CAFs can also originate from MSCs 107 and adipocytes 108 in breast cancer. The interaction between MSCs and CAFs is mainly studied in the context of their transformation. Osteopontin, derived from MDA-MB231 cells promotes the secretion of CCL5 by MSCs. These MSCs then chemotactically migrate to the tumor site and differentiate into CAFs, exhibiting increased expression of CAFs markers, such as α-smooth muscle actin (α-SMA), tenascin-c, and fibroblast-specific protein-1 109 . Furthermore, within the same cell line, it has been found that osteopontin facilitates the conversion of MSCs into CAFs in a MZF1/TGF-β1-dependent manner 110 . Inflammatory cytokines like TNF-α and IL-1 β are also responsible for the differentiation of MSCs. After several days of stimulation, MSCs adopt a CAF-like morphology and express high levels of α-SMA. These CAF-like cells secrete CCL2 and CCL5 cytokines, which stimulate chemokine receptors CCR2, CCR5, and CXCR1/2, as well as Ras-activating receptors expressed by MCF-7 and T47D cancer cells. Ultimately, this results in increased migration of cancer cells 107 . Based on these findings, it can be inferred that when infiltrating the TME, MSCs are exposed to a cascade of signaling factors released by tumor cells, potentially augmented by direct cell-cell interactions. Consequently, this exposure induces a loss of MSC stemness and prompts their differentiation into CAFs. The transformation into CAFs thus prevents MSCs from suppressing tumor cells and; instead, it promotes tumor progression. CAFs can facilitate breast cancer cells proliferation, invasion, metastasis, and drug resistance through various mechanisms111,112.
Because of the intricate connection between MSC and CAF, they also share certain similarities in the regulation of breast cancer. CAFs secrete some factors that are also produced by MSCs. Specifically, IL-6 and stromal cell-derived SDF-1 are secreted by both CAFs and MSCs, and these cytokines can promote the proliferation of breast cancer cells113–116. In addition, it has been observed that both CAFs and MSCs had a comparable impact on drug sensitivities. They were found to increase the effectiveness of the drug RAF265 (RAF inhibitor) on MDA-MB-231 cells by reducing ERK1/2 phosphorylation, indicating that, in most cases, CAFs differentiated from MSCs can simulate the drug sensitization effect of MSCs 117 .
Interactions Between MSCs and BCSCs
CSCs are identified as a group of cells possessing the capacity for self-renewal and the developmental capability to regenerate all the cell types found within a specific tissue 118 . Initially, mouse BCSCs were characterized by the expression of EpCAM+/CD44+/CD24−/low markers. Subsequently, human BCSCs were identified by isolating cells with a CD44+/CD24–/low/Lineage– expression phenotype 119 . Basal-like tumors display a predominant expression of CD44 and CD24, while HER2-positive and basal-like tumors have the highest expression levels of aldehyde dehydrogenase 1 (ALDH1), another stem cell marker 120 . The presence of stem cell markers, specifically ALDH1, has been associated with poor prognosis among women with breast cancer 121 .
MSCs express the stem cell marker ALDH1 and engage in interactions with cancer cells to control the self-renewal of BCSCs. IL-6 has been established as a direct modulator of BCSCs self-renewal, with its regulatory effect is mediated by the IL-6 receptor/GP130 complex, triggering the activation of STAT3 59 , 122 . Specifically, ALDH1-positive MSCs play a vital role in BCSCs through a cytokine feedback mechanism. Breast cancer SUM159 cells secrete IL6, which binds to IL6R and gp130 receptors expressed on ALDH1-positive MSCs, thereby facilitating the directed migration of MSCs toward primary tumor growth locations. Furthermore, MSC-derived CXCL7 interacts with cancer cells via the CXCR2 receptor, leading to the production of IL6 and IL8 59 . IL-8 also possesses the capacity to stimulate BCSCs self-renewal. Researchers identified significant expression levels of the IL-8 receptor CXCR1 in BCSCs through gene expression profiling 123 . In vitro, blocking CXCR1 had a specific and targeted effect on depleting the CSCs in SUM159 cells. In mouse xenografts, inhibiting CXCR1 resulted in a substantial reduction in BCSCs population, leading to diminished tumorigenicity and metastasis 123 .
It was noted that CSCs from MDA-MB-231 and T47D cell lines demonstrated a strong affinity for BM-MSCs. This interaction, mediated through CXCR4-dependent gap junctional intercellular communication (GJIC), resulted in elevated levels of Tregs, increased TGF-β levels, and a reduction in the Th17 immune response 124 . In addition, researchers discovered that MSCs facilitate the upregulation of miR-199a in breast cancer cells through direct contact. This upregulated miR-199a, in turn, suppresses the activity of forkhead-box P2 (FOXP2), a transcriptional regulator. The interaction between miR-199a and FOXP2 plays a crucial role in promoting CSC traits. Overexpressing miR-199a stably in various breast cancer cell lines, including MCF7/Ras, T47D, and MDA-MB-435, resulted in significant increases of BCSCs 125 . Interestingly, Sandiford et al. provide a comprehensive understanding of the intricate interplay between MSCs and CSCs during breast cancer dormancy in the BM perivascular region, involving the Wnt-β–catenin pathway. MSCs, as the primary niche cells encountered by breast cancer cells in the BM, exert direct and indirect effects on tumor cell behavior by releasing EVs. These EVs induce a specific Oct4alo subset of breast cancer cells to enter a state of quiescence and resistance to chemotherapy, ultimately leading to their dedifferentiation into BCSCs 126 .
The interactions between MSCs and CSCs are also reported in other tumors. In glioblastoma, it was observed that direct cellular interaction between UC-MSCs and glioblastoma stem cells elicited a notable decline in the growth rates of both cell populations. In contrast, the conditioned medium derived from UC-MSCs exhibited the capacity to stimulate CSC proliferation. This stimulatory effect was attributed to the activation of the ERR1/2 and Akt signaling pathways, potentially facilitated by the release of cytokines, including IL-8, GRO, ENA-78 (epithelial neutrophilactivating protein 78), and IL-6, by UC-MSCs 127 . Jean et al. provided evidence that co-culturing MSCs with glioblastoma stem cells facilitated the transfer of MSC-derived mitochondria to CSCs through tunneling nanotubes. This mitochondrial transfer event induced metabolic reprogramming and resistance to temozolomide of the glioblastoma stem cells 128 . In colorectal cancer, LoVo cells-derived IL-1 triggered release of PGE2 by MSCs, then PGE2 synergizing with IL-1 to stimulate MSCs secret IL-6, IL-8, and GRO-α. Moreover, MSC-derived PGE2 induced ALDHhigh CSCs by stimulating Akt/GSK-3/β-catenin signaling pathways 129 . Ma et al. declared that CD133+CD44+ colorectal cells were the most effective in recruiting MSCs compared with normal intestinal epithelial cells (NCM460), parental colon cancer cells and CD133—CD44— colon cancer cells. After been attracted, MSCs increased secretion of IL-8. IL-8 inhibitor can efficiently diminish the stem-like properties of CSCs and stimulate dormant CSCs to re-enter the cell cycle 130 . In lung cancer, IL-6 derived from AT-MSCs was found to augment the in vitro proliferation of LLC1 cells. Furthermore, it upregulated the expression of CSC markers in LLC1 cells, including pluripotent-related genes SOX2 and NANOG, as well as drug resistance-associated genes ALDH1A1 and ABCG2 131 . In gastric cancer, IL-8 produced by MSCs elevated the expression of PD-L1, leading to enhanced CSC-like characteristics in SGC-7901 cells, and ultimately fostering tumor progression 132 . In pancreatic cancer, BM-MSCs-derived exosomes circ_0030167 increased Wif1 expression while suppressing the Wnt8/β-catenin pathway through miR-338-5p, resulting in the suppression of the stem cell-like characteristics of pancreatic cancer cells and impedes the progression of tumors 133 .
Strategies eliminating BCSCs, including targeting the surface markers of BCSCs, disrupting BCSCs-dependent signaling pathways, promoting BCSCs differentiation, interfering with BCSCs metabolism, and modulating the TME. These approaches were extensively elucidated by Li et al. 134 in their study. Notably, owing to their precision in delivery and exceptional drug stabilization properties, nanodrugs have demonstrated significant promise in targeting BCSCs. Xie et al. developed an amphiphilic nanodrug consisting of all-trans retinoic acid (ATRA), the anti-cancer agent irinotecan (IRI), and the photothermal compound IR825. This nanoparticle not only exhibits remarkable antitumor efficacy in a 4T1 mouse model by promoting the differentiation of BCSCs into non-BCSCs but also allows for the monitoring of drug release through fluorescence and photoacoustic imaging 135 . Pan et al. engineered nanocomposite particles comprising GNSs (gold nanostars), dPG (dendritic polyglycerol), 3BP (3-bromopyruvate), TPP (triphenyl phosphonium), and HA (hyaluronic acid). These nanocomposites, using dPG as a platform, were designed to simultaneously inhibit the metabolism of BCSCs and precisely target them, resulting in the suppression of 4T1 tumor growth 136 .
Interactions Between MSCs and Breast Cancer Cells
MSCs have been observed to actively engage in tumor progression by establishing intricate communication networks with cancer cells. Notably, breast cancer cells recruit MSCs to create a favorable microenvironment that supports tumor cell proliferation, promotes EMT, facilitates invasion and migration, and even contributes to tumor dormancy.
The mechanisms by which MSCs enhance cancer cell proliferation are intricate and multifaceted. The co-injection of NLR-JIMT cells with patient-derived MSCs, specifically invasive cancer MSC-BRCA+/MSC-CA, and non-invasive cancer MSC-DCIS led to a significantly increased tumor volume of mice in comparison to co-injections MSC-H derived from healthy donors 137 . When MDA-MB231 cells were co-cultured with AT-MSCs, an increase in their viability as observed compared with cells cultured alone 81 . Notably, there was an upregulation in the activity of mammalian target of rapamycin (mTOR), a pivotal protein involved in cellular proliferation and viability. In addition, the levels of Beclin-1 and LC3 II, markers associated with autophagy, were decreased. These findings strongly suggest that MSCs promote cell proliferation in breast cancer by inhibiting the process of autophagy 81 . Co-culture of MCF-7 cells with MSCs results in the upregulation of SDF-1 gene expression, thereby promoting increased proliferation. Independent treatment of MCF-7 cells with SDF-1 alone also enhances their proliferation. Importantly, the inhibition of SDF-1 signaling through a CXCR4-specific inhibitor effectively mitigates the proliferative and migratory effects induced by MSCs in MCF-7 cells. These findings strongly suggest the involvement of the SDF-1/CXCR4 signaling pathway in mediating the cellular effects exerted by MSCs on MCF-7 cells 114 . Hypoxia, a prominent characteristic of solid tumors, stimulates the secretion of growth factors and cytokines, including TGF-β1, TGF-β2, and VEGF, by MSCs 138 . Moreover, it has been shown that TGF-β1 activates the SMAD signaling pathway, thereby promoting the proliferation of breast cancer cells 87 . MSCs can also modulating the growth of cancer cells through IL-6/STAT3 signaling pathway 139 . The Hippo signaling pathway, known for its role in tumorigenesis, has significant implications in breast cancer progression 140 . Exosomes secreted by hAT-MSC-differentiated adipocytes can stimulate MCF7 cells proliferation and migration by activating the downstream effectors YAP and TAZ of the Hippo pathway 141 .
MSCs influence the migration of MDA-MB-231 cells through secreting TGF-β, which activates several migratory proteins, including rho-associated kinase (ROCK), focal adhesion kinase (FAK), and MMPs 142 . Antonella et al. demonstrated that IL-6 and VEGF, secreted by MSCs, act as paracrine factors to enhance breast cancer cell migration. This enhancement is achieved by activating MAPK, AKT, and p38MAPK signaling pathways 143 . DDR2 is a specific collagen receptor tyrosine kinase primarily expressed in MSCs. It plays a role in ECM synthesis and wound healing 144 . In the context of human breast cancer metastasis, an intriguing observation emerges regarding the coordinated upregulation of DDR2 in both metastatic cancer cells and MSCs. This leads to the dysregulated activation of DDR2 signaling within breast cancer cells. Disrupting DDR2 in MSCs exerts a detrimental effect on their ability to induce DDR2 phosphorylation in breast cancer cells, thereby influencing the alignment, migration, and metastatic potential of tumor cells. Furthermore, mice lacking functional DDR2 exhibit reduced efficiency in the occurrence of spontaneous breast cancer metastasis 82 . In addition, TNFα-stimulated MSCs have a profound effect on breast tumor metastasis. They exhibit enhanced metastatic potential compared with N-MSCs, associated with increased expression of CCL5, CCR2, and CXCR2 ligands, along with enhanced infiltration of neutrophils within the TME. Subsequent interactions between neutrophils and tumor cells stimulate genes associated with metastasis, such as MMP-12, MMP-13, IL-6, and TGFβ, emphasizing the critical role of TNFα-activated MSCs in promoting tumor metastasis via the recruitment of CXCR2+ neutrophils 145 . Meanwhile, the presence of CCR5 in MDA-MB-231 cells, coupled with the secretion of CCL5 by MSCs, actively promotes cancer cell metastasis through the CCL5-CCR5 signaling pathway. Inhibition of CCR5 expression in MDA-MB-231 cells via shRNA knockdown and neutralization of CCL5 using a CCL5 monoclonal antibody effectively abolished the metastasis-enhancing effect of MSCs on MDA-MB-231 cells 48 .
During EMT transition, there is a notable downregulation of adhesion molecule, like E-cadherin, and the acquisition of mesenchymal markers, like N-cadherin and vimentin (VIM), leading to a shift toward a more mobile and invasive cell phenotype 146 . In breast cancer, EMT plays a pivotal role in facilitating tumor metastasis by enabling cancer cells to detach from the primary tumor, invade neighboring tissues, and establish secondary tumors at distant locations 147 . Furthermore, EMT is closely associated with the development of resistance to treatment, adding complexity to the management of breast cancer 148 . Co-culture of breast cancer cell lines with MSCs resulted in a significant increase in the expression of EMT-specific markers, including N-cadherin, VIM, Twist, and Snail, while the expression of E-cadherin protein was reciprocally decreased 149 . MSCs exert a notable influence on the behavior of human breast carcinoma cells by promoting the synthesis of lysyl oxidase (LOX). This, in turn, significantly enhances the metastatic potential of cancer cells toward the lungs and bones. The overexpression of LOX triggers EMT in MCF7/Ras cells 150 . In another co-culture setting with breast cancer cells, MSCs exhibit the capacity to produce TGF-β and IL-6 151 . TGF-β plays a pivotal role in modulating the initiation of EMT in MCF7 cells through the ZEB/miR-200 signaling pathway. The downregulation of TGF-β levels reverses EMT by inhibiting ZEB1/2 expression and enhancing the production of miR-200b and miR-200c 83 . These cytokines also upregulate the expression of long non-coding RNAs (LincK) in breast cancer cells. Notably, LincK induction triggers the progression of EMT in MCF-7, MDA-MB-453, and MDA-MB-231 cells. Suppression of LincK expression resulted in a notable decline in the proliferation, migration, and invasiveness of breast cancer cells both in vivo and in vitro, while overexpression of LincK had the opposite effects 151 . Notably, MSCs derived from invasive breast cancer patients demonstrated a greater ability to promote EMT compared with MSCs derived from healthy individuals or from non-invasive cancer patients 137 . Moreover, it has been reported that MSCs also modulate the EMT process through fusion with breast cancer cells. Pathophysiological fusion events involving MSCs have also been observed in gastric, ovarian, lung, and liver cancers152–156. Melzer et al. 157 observed altered Short Tandem Repeat (STR) profiles and upregulated EMT-related genes in the hybrid population of UC-MSCs and MDA-MB-231 cells. When GFP-tagged MSCs and mCherry-tagged MDA-MB-231 breast cancer cells were co-injected into mice, the spontaneous emergence of hybrid cells (constituting less than 0.5%) was observed within 14 days of tumor development, as indicated by the presence of cells displaying both fluorescent markers 158 . In vivo experiments revealed that hybrid cells exhibited accelerated tumor growth in mice compared with the original cancer cells, with an earlier onset of systemic metastasis 157 . The fusion between MSCs and breast cancer cells occurs within a few minutes in vitro. TNF-α and MMP-9 were found to stimulate cell fusion in breast cancer159,160.
Tumor dormancy refers to a state in which cancer cells enter a dormant or quiescent phase, characterized by their temporary arrest in growth and proliferation 161 . During this dormant period, tumor cells exhibit reduced metabolic activity and can remain undetectable for extended periods. Tumor dormancy poses a significant challenge in cancer treatment, as dormant cells have the potential to evade therapy and later reawaken, leading to disease recurrence and metastasis 162 . By employing 3D co-cultures to replicate cellular interactions within a developing TME, Thomas et al. observed the sequential encirclement of breast cancer cells by MSCs. This process facilitated the generation of cancer spheroids followed by internalization and degradation of MSCs by MDA-MB-231 cells. It enhanced the survival of nutrient-deprived breast cancer cells while simultaneously suppressing their tumorigenic potential, indicating a transition of the cancer cells into a dormant state 163 . Mechanically, NG2+/Nestin+ MSCs influence dormancy modulation by secreting TGFβ2 and bone morphogenetic protein 7 (BMP7), activating a quiescence pathway involving transforming growth factor-β receptor III (TGFBRIII) and bone morphogenetic protein receptor II (BMPRII), ultimately leading to p27 induction. Interestingly, ER+ breast cancer patients who did not experience systemic recurrence exhibited a greater occurrence of TGFβ2 and BMP7 detection in the BM 164 . Mohd Ali et al. 165 demonstrated that miR-941, found in the co-culture media of AT-MSCs and MDA-MB-231/MCF-7 cells, was associated with cancer dormancy and chemoresistance. miR-941 may inhibit the migration and invasion of breast cancer cells through the regulation of EMT, leading to increased E-cadherin expression and decreased VIM, SMAD4, and SNAI1 expression. Moreover, MSC-induced dormancy occurred following the spontaneous fusion of MSCs with MDA-MB-231 cells, resulting in the formation of MDA-MSC hybrid cells. In a mouse model, initially, these hybrid cells displayed a dormancy-like state. Approximately, 6 months later, these hybrid cells began generating tumors at a rate approximately 1.8 times faster than that of the MDA-MB-231 cells. RNA microarray analysis revealed a significant increase in the expression of dormancy-associated genes, including tumor necrosis factor receptor (TNFR), BMP1/7, TGF-β, and VEGF transcripts in the hybrid cells compared with the normal cancer cells 166 .
MSCs possess the capacity to differentiate into endothelial-like cells 167 . Recognizing the pivotal role of neo-angiogenesis in the regenerative medicine of MSCs as a viable means of repairing damaged tissue, recent efforts have concentrated on utilizing MSCs as a reservoir for generating ECs168,169. Wu et al. explored the potential of utilizing MSCs for breast reconstruction following mastectomy. In vitro findings demonstrated that the introduction of UC-MSCs promoted the proliferation, migration, and angiogenesis of human umbilical vein endothelial cells (HUVECs) through the integrin β1/ERK1/2/HIF-1α/VEGF-A signaling pathway. In vivo experiments revealed that UC-MSCs underwent differentiation into adipocytes and vascular ECs within the AT. Moreover, the group receiving UC-MSCs transplantation exhibited increased AT and a diminished presence of M1 macrophages compared with the control group in the transplanted fat 170 . In breast cancer microenvironment, there remains a debate regarding the facilitation of angiogenesis by MSCs. The co-culture of MCF-7 cells with MSCs or the addition of VEGF has been shown to enhance the formation of capillary-like structures by MSCs. Conversely, when MSC cells were treated with the conditioned medium of MCF-7 cells alone, they failed to form capillary-like structures. This underscores the importance of tumor cells and their secretion of VEGF in the recruitment of MSCs and the formation of abnormal vascular 171 . In contrast to the injection of 4T1 cells alone into nude mice, the co-injection group of 4T1 cells and BM-MSCs demonstrated a larger tumor size, an increased blood vessel area within the tumor, and reduced central necrosis. In addition, the expression levels of angiogenesis markers, including TGF-β, VEGF, and IL-6, were elevated, indicating that MSCs have the potential to enhance angiogenesis in breast cancer 172 . Li et al. 173 suggested that MSCs may promote angiogenesis in breast cancer by modulating the NOTCH1 signaling pathway. Nevertheless, it has been reported that MSCs-derived exosomes transport microRNA-100 to breast cancer cells, exerting a suppressive effect on angiogenesis by modulating the mTOR/HIF-1α/VEGF signaling axis 174 .
The Role of MSCs in the Treatment of Breast Cancer
Effects of MSCs on Chemotherapy
MSCs demonstrate inherent resistance to chemotherapy. When exposed to cisplatin, MSCs undergo senescence rather than apoptosis 175 . The resistance of breast cancer to chemotherapy, mediated by MSCs, is often associated with the induction of a “stemness” phenotype. Direct co-culture of ER+ breast cancer cells with MSCs results in a remarkable transformation, acquiring characteristics resembling CSCs. This acquisition of CSC traits is accompanied by an increased resistance to tamoxifen and fulvestrant, two commonly used hormonal therapies for breast cancer 176 . Preconditioning MSCs with cisplatin elicits the secretion of various cytokines, including IL-6, CXCL1, and IL-8, and induces notable alterations in the phosphorylation patterns of PLC-y1, WNK1, RSK1/2/3, p53, and c-Jun. Consequently, this leads to enhanced resistance to chemotherapy and an augmented stemness observed in breast cancer cell lines both in vitro and in vivo experimental models 175 . Apart from their intrinsic resistance to chemotherapy-induced apoptosis, MSCs have the ability to protect MCF-7 cells from cisplatin-induced apoptosis by secreting IL-6 and activating the IL-6/STAT3 signaling pathway in cancer cells 177 . The role of IL-6 in the development of acquired chemoresistance in breast cancer is significant. Doxorubicin-sensitive breast cancer cells lack IL-6 expression, whereas doxorubicin and PTX-resistant breast cancer cells exhibit elevated IL-6 production, along with upregulated expression of the multidrug resistance gene mdr1, as well as increased levels of the transcription factors C/EBPβ and C/EBPδ 178 . In addition, the release of CXCL1 by MSCs leads to an upregulation of ABCG2, a multidrug transporter associated with chemoresistance, and plays a significant role in conferring resistance to doxorubicin in TNBC cells 179 . MSC-derived IL-8 also plays a role in upregulating the expression of ABCG2, leading to decreased doxorubicin sensitivity in MDA-MB-231 cells 180 . Another study has revealed that MSCs stimulate the upregulation of C-terminal Src kinase-binding protein (PAG1/Cbp), which, in turn, activates Src, facilitating resistance against adriamycin hydrochloride 181 .
However, there is also evidence suggesting that MSCs can enhance the sensitivity of breast cancer cells to chemotherapy. SLC9A1 functions as a positive regulator of the Wnt/β-catenin signaling pathway, which is associated with breast cancer cell proliferation and resistance to the chemotherapy. The transfer of miR-1236 by AT-MSC-derived exosomes results in the suppression of SLC9A1 mRNA expression, leading to decreased proliferation and increased cisplatin sensitivity in breast cancer cells 182 . Kucerova et al. concluded that the soluble factors present in the conditioned media of MSCs or in the direct co-culture of breast tumor cells with AT-MSCs do not confer chemoresistance. Intriguingly, the presence of AT-MSCs resulted in a significant increase in the susceptibility of SKBR3 tumor cells to the cytotoxic effects of doxorubicin and 5FU 90 . Overall, the reciprocal interactions between tumors and MSCs not only shape the biological characteristics of the tumor as a complex entity but also influence its response to chemotherapy. The impact of MSCs on the response of tumor cells to chemotherapy is multifaceted and can be influenced by the tumor cell’s state and the characteristics of specific MSC populations.
Effects of MSCs on Radiotherapy
Radiation therapy plays a pivotal role in the multimodal approach to breast cancer management, including early-stage, locally advanced, and metastatic breast cancers 183 . After breast-conserving surgery, administering radiotherapy to the preserved breast significantly reduces the recurrence rate of the disease and lowers the breast cancer mortality rate by approximately one-sixth 184 . Radiation therapy stimulates the release of inflammatory substances that facilitate the recruitment of MSCs into the TME. Within 48 h of radiation exposure, 4T1 breast carcinomas that received 2Gy exhibited a 34% higher level of MSC integration compared with the untreated side. Similarly, in vitro experiments demonstrated that MSC migration toward conditioned media from irradiated 4T1 cells increased by 50%–80% compared with control group. Mechanically, it was discovered that irradiation of tumor cells led to an upregulation of the chemokine receptor CCR2 in MSCs. Importantly, inhibiting CCR2 significantly reduced the migration of MSCs 185 . Capitalizing on this characteristic, Steven et al. investigated the potential of utilizing radiation to increase the recruitment of MSCs within the TME. They demonstrated that upon intravenous infusion, MSCs were detected within subcutaneous tumors as early as 3 days after administration, with minimal or undetectable migration to other tissues. Lentivirus-transduced MSCs exhibited effective and consistent transgene expression. In addition, the number of MSCs homing to tumors can be increased by administering radiation therapy to the tumors prior to MSC infusion with no difference in tumor size 65 . These results suggest that through genetic modification of MSCs, radiotherapy can enhance the effectiveness of MSC-based gene therapy, thereby improving therapeutic outcomes. Accordingly, Zhang et al. engineered BMSCs to serve as carriers capable of delivering both early growth response protein 1-linked human sodium iodide symporter (Erg1-hNIS) and gold nanoparticles (AuNCs) simultaneously. The findings revealed a synergistic effect of the combination treatment involving BMSC-Egr1-hNIS+AuNCs and radiation therapy in mice with MDA-MB-231 tumor xenografts 186 .
While there is evidence suggesting that radiation therapy can increase the chemotactic response of MSCs and has been employed in therapeutic approaches, it is important to recognize that radiation therapy can also induce changes in the biological properties of MSCs. In addition, MSCs have been implicated in altering the sensitivity of breast cancer to radiotherapy. Anne et al. conducted an analysis of wound fluid collected from breast cancer patients who underwent breast-conserving surgery. Among the patients, 21 individuals received intraoperative radiotherapy consisting of a single dose of 20 Gy (referred to as the IORT group), while 21 patients underwent surgery without receiving intraoperative radiotherapy (referred to as the control group). Remarkably, the wound fluid obtained from the IORT group exhibited significant inhibitory effects on various functions of MSCs, such as proliferation, wound healing, and migration. Furthermore, it notably downregulated the expression of VEGF and growth-related oncogene α (GROα) by MSCs compared with the control group 187 . In the context of obesity, AT-MSCs have been implicated in promoting radiation resistance in breast cancer cells. This is achieved by upregulating the production of leptin, which, in turn, induces the secretion of IL-6 and activates NOTCH signaling pathways 188 . MSCs can also secret insulin-like growth factor 1 (IGF-1) to promote radiation resistance in breast cancer cells 189 . However, studies have demonstrated that treatment with MSC-conditioned medium not only reduces the growth of MDA-MB-231 cells but also enhances the sensitivity of these cancer cells to radiation therapy by inhibiting the activation of the Stat3 signaling pathway 89 .
Effects of MSCs on Immunotherapy and Target Therapy
Historically, breast cancer has been recognized as a tumor with limited immunogenicity and a lower frequency of somatic mutations compared with malignancies, such as melanoma and lung cancer 190 . However, TNBC has shown greater responsiveness to programmed cell death protein 1 (PD-1) inhibitor pembrolizumab 191 . At present, there is a lack of relevant research elucidating the impact of MSCs on the treatment of breast cancer with immune checkpoint inhibitors (ICIs). But there is evidence indicating that MSCs can modulate the expression of PD-1/PD-L1 of breast cancer cells. For example, MSCs induce the expression of PD-L1 in MCF-7 cells through the secretion of CCL5 192 . Exosomes derived from MSCs attenuate antitumor T cell responses by upregulating PD-L1 expression in M2 macrophages and increasing the PD-1 levels of T cells 95 . In addition to ICIs, in a co-culture experiment involving CD19 CAR-T cells, Pfeiffer cells, and M2 macrophages, Rui et al. demonstrated that after 24 h of exposure, CAR-T cells killed 67% of Pfeiffer cells, and the cell-killing activity increased to 93% after 48 h compared with the control group. Nevertheless, the introduction of MSC into the co-culture noticeably suppressed the cytotoxic potential of CAR-T cells 102 . Based on these findings, we hypothesize that MSCs may potentially counteract the efficacy of breast cancer immunotherapy.
In terms of targeted therapy, trastuzumab was initially approved for the treatment of HER-2 positive advanced breast cancer. However, resistance to trastuzumab has posed a significant challenge in treating patients effectively 193 . Jing et al. co-cultured MSCs with HER2-positive SKBR-3 and BT474 breast cancer cells and observed that MSC co-culture induced resistance to trastuzumab. Trastuzumab treatment significantly suppressed tumor growth in BALB/c nude mice implanted with SKBR-3 cells. However, this effect was significantly reversed when SKBR-3 were experimentally manipulated to overexpress LncRNA AGAP2-AS1. Clinical samples indicated that circulating AGAP2-AS1 levels serve as a predictive factor for the response of breast cancer patients to trastuzumab treatment. Mechanistically, the AGAP2-AS1, derived from MSC, promotes stemness and confers resistance to trastuzumab treatment by regulating fatty acid oxidation (FAO) and the HuR/miR-15a-CPT1 axis in breast cancer cells 194 . In addition, the physical interaction between MSCs and breast cancer cells contributes to trastuzumab resistance by activating Src/PI3K/AKT signaling pathway. MSCs’ presence also results in increased HER2 expression and decreased PTEN levels, indicating MSCs’ regulatory role in mediating the functional interaction between HER2 and PTEN through Src/PI3K/AKT activation 195 . Next, lapatinib is a dual tyrosine kinase inhibitor that selectively targets and inhibits epidermal growth factor receptor (EGFR/ErbB1) and HER2/ErbB2 196 . Sarkis et al. observed that the expression of PEAK1 in MSCs provides protection to BT474 and MCF7 cells against lapatinib-induced cytotoxicity. They demonstrated a novel PEAK1-INHBA/activin-A-dependent pathway in MSCs that plays a crucial role in inducing resistance to lapatinib in HER2-positive breast cancer cells. In addition, they identified that MSCs expressing PEAK1 contribute to the emergence of distinct subpopulations within HER2-positive breast cancer cells characterized by high levels of p-Akt, MCL1, and GRP78, as well as low levels of p-γH2AX and VIM. These subpopulations exhibit resistance to lapatinib and demonstrate enhanced tumorigenic potential both in vitro and in vivo settings 197 .
The Therapeutic Potential of MSCs for Breast Cancer
The remarkable homing capabilities of MSCs to tumor sites have been harnessed as a valuable approach for efficiently and selectively delivering therapeutic agents (Table 2).
Therapeutic Potentials of MSCs in Breast Cancer.

GNR@HPMOs-PTX, gold nanorod-embedded hollow periodic mesoporous organosilica nanospheres; EV, extracellular vesicle; WJ, Wharton’s jelly; BM, bone marrow; AT, adipose tissue; UC, umbilical cord; DP, dental pulp.
First, researchers have engineered MSCs to produce antitumor factors, endowing them with the ability to selectively target and eliminate breast cancer cells. Through genetic modifications involving Lrp5, β-catenin, Snail, or Akt, MSCs acquired the ability to suppress the growth of breast cancer cells, these modifications were accompanied by an increase in the expression of Hsp90ab1, calreticulin as well as a decrease in the levels of CXCL2 and PD-L1. In vivo experiments demonstrated that administering the conditioned medium significantly inhibited the tumor growth and decrease bone metastasis 198 . In another study, AT-MSCs were genetically modified through retroviral transduction with either cytosine deaminase::uracil phosphoribosyltransferase (CD:UPRT) or Herpes simplex virus thymidine kinase (HSVtk). In vitro studies showed that a combination of CD:: UPRT-MSC and HSVtk-MSC had a synergistic cytotoxic effect on MDA-MB-231 cells. When administered systemically, this approach effectively inhibited the migration of tumor cells to the lungs of mice 199 . AT-MSCs were also transfected with TGF-β1, which significantly inhibited the development of MCF-7 and MDA-MB-231 cells. The conditioned medium of TGF-β1/MSC were found to stimulate the SMAD signaling pathway, resulting in an increase in pSMAD2/3 expression and a decrease in SMAD4 expression in breast cancer cells 200 . Chen et al. modified MSCs with IL-12 and explored its effect on breast cancer therapy. They demonstrated that IL-12-engineered MSCs (IL-12/MSCs) can migrate to tumor sites and effectively suppress tumor growth and metastasis without systemic toxic effects. This was in contrast to free IL-12, which only partly retards tumor growth and had obvious toxic effects due to nonselective expression 201 . Dwyer et al. investigated the utility of MSCs expressing the sodium iodide symporter (NIS) for both imaging and therapeutic purposes in breast cancer. They found that MSC-NIS injection allowed for non-invasive tracking of MSC migration and transgene expression, resulting in a significant decrease in tumor growth 203 . Giulia et al. introduced human tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) into AT-MSCs using a retroviral vector. These TRAIL-armed AT-MSCs successfully targeted different types of tumor cell lines, including cervical, pancreatic, and colon neoplasms in vitro. Importantly, they demonstrated antitumor efficacy against BT549 breast cancer cells that were resistant to TRAIL treatment 204 .
Second, MSC-derived EVs can be used as a means of delivering drugs or microRNAs to tumor cells. This method offers an advantage over traditional drug delivery methods because MSCs can efficiently and selectively deliver drugs to specific locations. MSC-derived exosomes loaded with taxol exhibit efficient tumor-targeting properties and reduced side effects. In vitro testing on human cancer cells showed 80-90% cytotoxicity. Notably, compared with achieving the same cytotoxic results using pure taxol, the concentration of taxol in MSC exosomes was 7.6 times lower. Furthermore, in vivo studies on mice bearing highly metastatic MDA-hyb1 breast tumors revealed significant therapeutic effects of MSC taxol exosomes 206 . Wu et al. developed a strategy in which MSCs were loaded with gold nanorod-embedded hollow periodic mesoporous organosilica nanospheres (GNR@HPMOs) with paclitaxel (PTX), named GNR@HPMOs-PTX@MSCs. In vitro and in vivo experiments have demonstrated that this approach has a synergistic effect on killing breast cancer cells and inhibiting tumor growth 207 . O’Brien et al. engineered MSCs to secrete EVs enriched with a tumor-suppressing microRNA-379 (miR-379). While the introduction of MSC/miR-379 cells did not influence the growth of T47D tumors, systemic administration of EVs enriched with miR-379 demonstrated pronounced therapeutic efficacy 208 . EVs isolated from CD90low AT-MSCs demonstrated antitumor activity, and miR-16-5p loaded into CD90low AT-MSC-EVs enhanced apoptosis in E0771 and 4T1 tumor cells, slowing tumor growth both in vitro and in vivo 80 . Of note, E1A modified MSCs were used to delivery CD3-HAC, a bifunctional protein consisting of an anti-CD3 single-chain variable fragment (scFv) and a high-affinity consensus (HAC) domain. MSCs were able to selectively migrate to metastatic sites of breast cancer and secreting CD3-HAC proteins within the TME. Importantly, when combined with 5-FU treatment, MSC-CD3-HAC-E1A therapy effectively suppressed MDA-MB-231 growth in mice 209 .
In addition, we conducted a search on the ClinicalTrials.gov website to determine if any relevant clinical trials had been conducted. Unfortunately, our search yielded no results regarding trials directly addressing the application of MSCs for breast cancer treatment. However, we did identify trials investigating the use of MSCs in other types of cancer, which we have summarized in Table 3.
Clinical Trials Investigating the Use of MSCs in Cancer Treatment.

UC, umbilical cord; AT, adipose tissue; BM, bone morrow; NIS, sodium iodide symporter; CD, cytosine deaminase; Ad5-DNX-2401, adenovirus DNX-2401; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Conclusion
The role of MSCs in the breast TME is significant and multifaceted. MSCs are recruited to breast tumor sites through molecular signals released by damaged tissue, similar to the process of wound healing. Once within the TME, MSCs interact with various cell populations, including immune cells, CAFs, CSCs, and cancer cells. MSCs exhibit the ability to modulate immune responses and create an immunosuppressive environment, thereby influencing the tumor immune landscape. In addition, their interaction with CAFs and CSCs contributes to the promotion of tumor progression and self-renewal capabilities. Furthermore, MSCs establish communication networks with cancer cells, affecting tumor cell behavior and characteristics, such as proliferation, migration, EMT, and dormancy. The complex interplay between MSCs and the components of the breast TME highlights the need for further investigation. In most cases, MSCs play roles in breast cancer therapeutic resistance, but there is also evidence indicating their abilities to sensitize cancer cells to chemotherapy and radiotherapy.
Furthermore, exploring the potential of MSCs as a therapeutic tool is an exciting avenue of research. MSCs possess inherent regenerative and homing properties, making them attractive candidates for cell-based therapies. MSCs can be engineered to express therapeutic molecules or deliver anti-cancer agents directly to tumor sites, further enhancing their efficacy. However, challenges, such as optimizing MSC dosing, timing, and delivery routes, as well as ensuring their safety, need to be addressed for successful clinical translation. Moreover, the integration of MSC-based therapy with complementary treatment modalities, such as chemotherapy, radiation therapy, or immunotherapy holds immense potential for achieving synergistic effects and enhancing treatment outcomes.
In summary, unraveling the intricate relationship between MSCs and the breast TME has the potential to uncover novel therapeutic targets and advance our understanding of breast cancer biology. Continued research efforts in this field will pave the way for innovative treatment approaches and personalized interventions that can improve patient outcomes.
Footnotes
Acknowledgements
Not applicable.
Author Contributions
Not applicable.
Availability of Data and Materials
Not applicable.
Ethical Approval
Not applicable
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article, and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.