
Abstract
Asthma is a complex and heterogeneous disease characterized by chronic airway inflammation, airway hyperresponsiveness, and airway remodeling. Most asthmatic patients are well-established using standard treatment strategies and advanced biologicals. However, a small group of patients who do not respond to biological treatments or are not effectively controlled by available treatment strategies remain a clinical challenge. Therefore, new therapies are urgently needed for poorly controlled asthma. Mesenchymal stem/stromal cells (MSCs) have shown therapeutic potential in relieving airway inflammation and repairing impaired immune balance in preclinical trials owing to their immunomodulatory abilities. Noteworthy, MSCs exerted a therapeutic effect on steroid-resistant asthma with rare side effects in asthmatic models. Nevertheless, adverse factors such as limited obtained number, nutrient and oxygen deprivation in vitro, and cell senescence or apoptosis affected the survival rate and homing efficiency of MSCs, thus limiting the efficacy of MSCs in asthma. In this review, we elaborate on the roles and underlying mechanisms of MSCs in the treatment of asthma from the perspective of their source, immunogenicity, homing, differentiation, and immunomodulatory capacity and summarize strategies to improve their therapeutic effect.
Introduction
Asthma is a chronic inflammatory airway disease characterized by airway hyperresponsiveness (AHR), reversible airflow restriction, and airway remodeling 1 . It affects more than 350 million people worldwide and its prevalence and mortality rate are increasing annually 2 . Its main pathological characteristics include the infiltration of inflammatory cells into the airway wall, plasma extravasation, mucosal edema, goblet cell metaplasia, excessive subepithelial collagen deposition, gland hypertrophy, smooth muscle cell hyperplasia/hypertrophy, and angiogenesis3–5. Imbalances in helper T lymphocyte type 1 (Th1)/Th2 and Th17/regulatory cells (Treg), mast cell (MC) degranulation, antigen presentation by dendritic cells (DCs), and inflammatory factors and mediators [e.g., interleukin (IL)-4, IL-5, IL-13, IL-17, and IL-9] released by inflammatory cells such as M1 macrophages, eosinophils, and neutrophils are involved in mediating pathological changes in asthma 6 .
Recent advances have categorized asthma based on distinct molecular pathways (endotypes) and variable clinical symptoms (phenotypes)3,7. Asthma phenotypes are classified according to various factors such as disease severity, age at onset, symptom triggers, inflammatory patterns, frequency of exacerbations, and airflow obstruction. Meanwhile, asthma endotypes are categorized based on distinct pathophysiologic mechanisms at a cellular and molecular level 7 . It was subdivided into allergic and non-allergic eosinophilic asthma and non-eosinophilic asthma 8 . Eosinophilic asthma is dominated by type 2 cytokines and is known as Th2-high subtypes, whereas non-eosinophilic asthma is the absence of type 2–driven inflammation and is driven primarily by neutrophilic inflammation and is known as Th2-low subtypes8,9. Allergic eosinophilic asthma is characterized by early onset with an atopic background and elevated immunoglobulin E (IgE) levels 8 . Allergen-specific Th2 cells produce type 2 cytokines, including IL-4, IL-5, and IL-13, which result in allergen-specific IgE production by B cells. This leads to the activation of eosinophils and MCs, causing lung tissue damage and ultimately increased airway hypersensitivity 9 . Non-allergic eosinophilic asthma is often later onset with normal IgE levels 8 . Non-eosinophilic asthma is characterized by persistent symptoms and airflow limitation but is poorly responsive to traditional corticosteroid therapy, and has normal sputum and peripheral blood eosinophil counts7,8. It often occurs in association with obesity or neutrophilic predominance in the airways 7 .
Most asthma patients respond well to conventional drugs, such as anti-leukotrienes, anti-cholinergics, short- and long-acting beta-2 agonists, and inhaled corticosteroids 10 . However, 5%–10% of asthma symptoms remain uncontrolled even after receiving conventional treatment. Refractory asthma treatment costs represent approximately 50% of all asthma cases and have a significant financial impact on individuals, families, and society as a whole 10 . Moreover, these drugs provide relief from asthma symptoms while not modifying the immune system. Biological immunotherapies have emerged as a promising personalized medicine approach for the treatment of severe asthma with examples including anti-IgE (omalizumab), anti-IL-5 (reslizumab), anti-IL-5 receptor (bevacizumab), anti-IL-4/IL-13 receptor (dupilumab), anti-IL-17 receptor (brodalumab), and anti-TSLP (tezepelumab) therapies8,9. However, the heterogeneity of asthma pathogenesis presents challenges in establishing stable treatments for individual patients. Moreover, long-term use of these drugs also raises concerns about potential side effects which may limit their application. Therefore, there is an urgent need to discover new, safe, and effective alternative therapeutics.
Mesenchymal stem cells (MSCs), according to the minimal criteria definition, are plastic adherent cells with the potential for trilineage differentiation (osteogenic, adipogenic, and chondrogenic) and specific cell surface marker expression (CD90, CD105, and CD73) but lack the expression of CD45, CD34, CD14, CD79, and human leukocyte antigen (HLA) DR 11 . It can be derived from various tissues including bone marrow (BM), umbilical cord (UC), adipose tissue (AT), placenta (PL), dental pulp (DP), Wharton’s jelly (WJ), and lungs12–14. Alternatively, they can be differentiated from induced pluripotent stem cells (iPSCs) which are derived from terminally differentiated human cells that have undergone reprogramming to attain a pluripotent state 15 . Due to the easy accessibility, few ethical issues, low immunogenicity, multipotency, and immunomodulatory attributes, MSCs have received significant attention in the treatment of immune-mediated disorders 12 , including asthma. Accumulating evidence showed that following systemic transplantation, MSCs were swiftly trapped in the pulmonary vascular bed as a result of their comparatively large size 16 . Moreover, MSCs can alleviate airway inflammation, reduce excessive mucus production and hyperresponsiveness, as well as repair damaged epithelial cells, and relieve airway obstruction. Noteworthy, they are capable of suppressing airway goblet cell proliferation, subepithelial collagen deposition, and fibrosis which effectively ameliorates or even reverses the process of airway remodeling17–19. Furthermore, in preclinical trials, MSCs have shown satisfied therapeutic potential for steroid-resistant and chronic asthma20,21. However, MSC-based strategies for asthma treatment are challenging. For example, limited in vitro amplification, gradual loss of stemness, and harsh microenvironments impair the therapeutic potential of transplanted MSCs and impede their prospects for clinical application22–25. Over the years, various strategies have been used to improve therapeutic effects by enhancing the properties of MSCs or the local microenvironment to help MSCs survive26–30.
This review elaborates on the mechanisms related to MSC-based therapy in asthma and summarizes strategies to improve the efficacy of MSCs, thus providing a reference for stem cell therapy in asthma.
MSC-Based Therapy for Asthma
Sources
MSCs can be isolated from almost all tissues 12 . BM-MSCs were the first to be isolated. Intratracheal administration of mouse-BM-MSCs is more effective than the administration of mouse mouse-lung-MSCs in reducing lung inflammation and increasing anti-inflammatory mediators in an allergic asthma model 31 . However, mouse-lung-MSCs show higher expression of several basement membrane proteins and growth factors, which makes them persist longer than mouse-BM-MSCs in injured lung tissue after systemic administration 32 . Moreover, human-BM-MSCs exert better capabilities than human-AT-MSCs in relieving airway inflammation, AHR, and remodeling in chronic asthma 18 . However, BM-MSCs are limited by the small amounts and their low expansion potential, along with invasive aspiration procedures 33 . Human-UC-MSCs are easily harvested using noninvasive methods and are obtained in abundant quantities while preserving long passages 34 . Moreover, human-UC-MSCs show higher immunosuppressive capacity than human-BM-MSCs as they are characterized by increased levels of immunomodulatory surface protein and cytokines such as IL-1β, IL-8, CD200, and CD274 34 . Human AT-MSCs exert more potent immunomodulatory effects than human-BM-MSCs and suppress the proliferation of stimulated peripheral blood mononuclear cells (PBMCs), inhibiting the differentiation of monocyte-derived immature DCs and secreting higher levels of immunomodulatory cytokines, including IL-6 and transforming growth factor-β1 (TGF-β1) 35 . Human DP-MSCs and human-WJ-MSCs are more effective than human-AT-MSCs or human-BM-MSCs in inhibiting mtROS levels in stress receptor cells 36 . Human iPSC-MSCs as a novel cell type display a higher proliferation capacity and telomerase activity, as well as lower immunogenicity compared with human-BM-MSC, which have attracted much attention21,37. Human urine–induced pluripotent stem cell-derived MSCs (U-iPSC-MSCs) possess a great proliferation capacity in vitro without senescence even at passage 50 21 . However, the amazing proliferative capacity of iPSC-MSCs raises concerns about their potential risk of tumorigenesis38,39.
Collectively, the distinct characteristics and secretion profiles of MSCs derived from different tissues may lead to distinct therapeutic effects in asthma. It is necessary to further study the mechanism of MSCs in the treatment of asthma to choose the optimal MSCs to maximize the therapeutic effect of MSCs in asthma.
Low Immunogenicity
The low immunogenicity of MSCs is one of the most significant advantages of MSCs for asthma treatment. MSCs express low levels of class I major histocompatibility complex molecules and lack class II major histocompatibility complex and co-stimulatory molecules (e.g., CD40, CD40 ligand, CD80, and CD86) enabling transplanted MSCs to evade immune surveillance and across major histocompatibility barrier40,41. Moreover, MSCs secrete a series of immune-inhibitory molecules (e.g., HLA-G and IL-10) involved in MSC-induced immunosuppression 42 . Numerous preclinical trials have confirmed that the application of allogeneic MSCs for different types of asthma has a high safety profile and significant therapeutic effects have been achieved (Table 1). Furthermore, numerous clinical trials have confirmed the safety of donor MSCs in various diseases43–45.
MSC-Based Therapy for Several Types of Asthma.

Mus: mouse; IT: intratracheal; IV: intravenous; IN: intranasal; OVA: ovalbumin; BALF: bronchoalveolar lavage fluid; AHR: airway hyperreactivity; ILC2s: innate lymphoid cells group 2; DCs: dendritic cells; MLN: mediastinal lymph nodes; AwCT: airway contractile tissue; ECM: extracellular matrix; Ig: immunoglobulin; BM-MSCs: bone marrow–derived mesenchymal stem cells; UC-MSCs: umbilical cord–derived mesenchymal stem cells; AT-MSC: adipose tissue–derived mesenchymal stem cells; iPSC-MSCs: induced pluripotent stem cells (iPSC)-derived mesenchymal stem cells; MCA-MSC: induced pluripotent stem cell and mesenchymoangioblast–derived mesenchymal stem cells; PL-MSCs: placental-derived mesenchymal stem cells; LTB4: leukotriene B4; Cys-LT: cysteinyl leukotriene; COX-1: cyclooxygenase-1; Cytb: cytochrome b; ND-1: NADH-ubiquinone oxidoreductase chain 1; Nrf2: nuclear factor erythroid2–related factor 2; IFN-γ: interferon-γ; TNF-α: tumor necrosis factor α; HDM: house dust mite; Der f: house dust mite; DEP: diesel exhaust particle; Poly I: C: polyinosinic-polycytidylic acid; LPS: lipopolysaccharide; H2O2: hydrogen peroxide; Treg: regulatory cells; WBC: white blood cell; TGF-β: transforming growth factor-β; RORγ: retinoic acid receptor–related orphan receptor γ; Foxp3: forkhead box protein p3; PAS: periodic acid-Schiff; MMP-9: matrix metalloproteinase-9; FGFb: basic fibroblast growth factor; Gata3: GATA binding protein 3; RANTES: reduced upon activation, normal T cell expressed and secreted; VCAM-1: vascular cell adhesion molecule-1; ICAM-1: intercellular cell adhesion molecule-1; Fgf-1: fibrosis growth factor-1; Fn-1: fibronectin-1; MoMs: SiglecFCD11c-CD11b+ monocyte-derived macrophages; Muc5ac: mucin 5ac; Arg1: Arginase 1; Retnlα: resistin like α; AMs: alveolar macrophages; Rorc: RAR-related orphan receptor C.
Homing
MSCs possess a strong migratory potential owing to their particular components with cytoskeletal protein filaments (mainly microtubules, actin filaments, and intermediate filaments) and many parallel-arranged slender myofilaments and filamentous pseudopods around the cell periphery 67 . Owing to their migratory ability and inflammatory chemotaxis capacity, transplanted MSCs migrate to the damaged tissue and secrete various factors with powerful immunomodulatory effects to play a role in repair68,69.
MSC homing is a coordinated multistep process involving cytokines, chemokines, adhesion molecules, and matrix metalloproteinases 69 . Chemokine receptors [such as chemokine receptor 7 (CXCR7), CXCR4, C-C motif chemokine ligand 2 (CCL2)], adhesion molecules [e.g., vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1], and integrins (e.g., very late appearing antigen-4) on the surface of delivered MSCs interact with inflammatory factors released by effector cells and direct them from the circulation to damaged tissues for immunomodulation and tissue repair 69 . The expression of chemokines and adhesion factors varies across MSCs because of their heterogeneity.
In mouse models of allergic asthma, exogenous mouse-BM-MSCs migrate to inflamed sites depending on the stromal cell–derived factor-1a/CXCR4 axis. In contrast, CXCR4 inhibition decreases MSC migration to the lungs 70 . CXCR7 overexpression in rat-BM-MSCs significantly promotes rat-BM-MSC homing to damaged lung tissue 71 . In a cockroach allergen extract (CRE)-induced lung inflammation mouse model, CRE activated the aryl hydrocarbon receptor expressed on mouse-BM-MSCs and promoted the migration of mouse-BM-MSCs to the lungs 72 .
In addition to the surface molecules of MSCs, MSC homing relies on other cytokines during asthma. For instance, TGF-β1 released from the allergen-activated epithelium contributes to the recruitment of mouse-BM-MSCs toward the damaged airway, while fewer mouse-BM-MSCs migrate to the lungs using a TGF-β1-neutralizing antibody 72 . In particular, only 11.6% of the delivered mouse-BM-MSCs reached the inflamed area of the lung after systemic administration, implying a discount in the survival rate of the MSCs or low survival rates of the homing cells 63 . This could be attributed to the heterogeneity of MSCs, gradual loss of homing molecules during the amplification process, and harsh microenvironmental alteration23,73. Changing the culture conditions of MSCs to optimize the expression of homing molecules, modifying cell surface receptors using genetic engineering, or altering the target tissue microenvironment in advance can improve their homing efficiency.
Differentiation
MSCs can differentiate into multiple cell types and play an essential role in the repair of damaged lung tissue. For example, human UC blood-derived MSCs and rat-BM-MSCs differentiate into AEC2s in an epithelial cell medium for several days of culture74,75. Engrafted mouse-BM-MSCs homed to the injured bleomycin-induced lung and assumed lung cell phenotypes 76 . In the phosgene-induced acute lung injury rat model, alveolar epithelial type 2 cells (AEC2s)-specific marker prosurfactant protein C (red) were observed to coincide with donor rat-BM-MSC (green) signals in the lung tissues 24 h after MSC administration, indicating the probable differentiation of the rat-BM-MSCs into AEC2s 71 . However, Mariñas-Pardo et al. 60 failed to track MSCs differentiated into airway wall tissue in house dust mite (HDM)-induced asthma mouse models using a 2-week cutoff. The discrepancy among those results may be attributed to MSCs fusing with resident cells instead of differentiation. In addition, differential impacts of MSCs on a long-term and short-term basis also lead to divergent differentiation outcomes in vivo. For instance, Luan et al. 77 reported that, in a monocrotaline-induced pulmonary arterial hypertension mice model, after 6 months of rat-BM-MSC intravenous injection, co-localization of red fluorescence–positive rat-BM-MSCs and green fluorescence spots of vascular endothelial cells resulted in a yellow color within many lung regions, while the co-localization did not occur at 2 and 8 weeks after transplantation.
The underlying mechanism for MSC differentiation in asthma is complicated and is yet to be understood. Additional in vivo studies are needed to characterize the fate of MSCs and the impact of their persistence in asthma.
Immunomodulation
MSCs can downregulate local inflammatory responses, reduce inflammatory damage, and reverse airway remodeling by restoring the Th1/Th2 balance 10 , reverse the Th17/Tregs imbalance 52 , inhibit DC maturation and antigen presentation 78 , suppress the degranulation response of MCs 79 , restrain innate lymphoid cell group 2 (ILC2)-mediated pathology 80 , promote the shift of M1 macrophages to M2 macrophages 57 , induce the differentiation of Treg cells 81 , and repair epithelial injury 19 through paracrine secretion or direct cell–cell contact (Fig. 1).
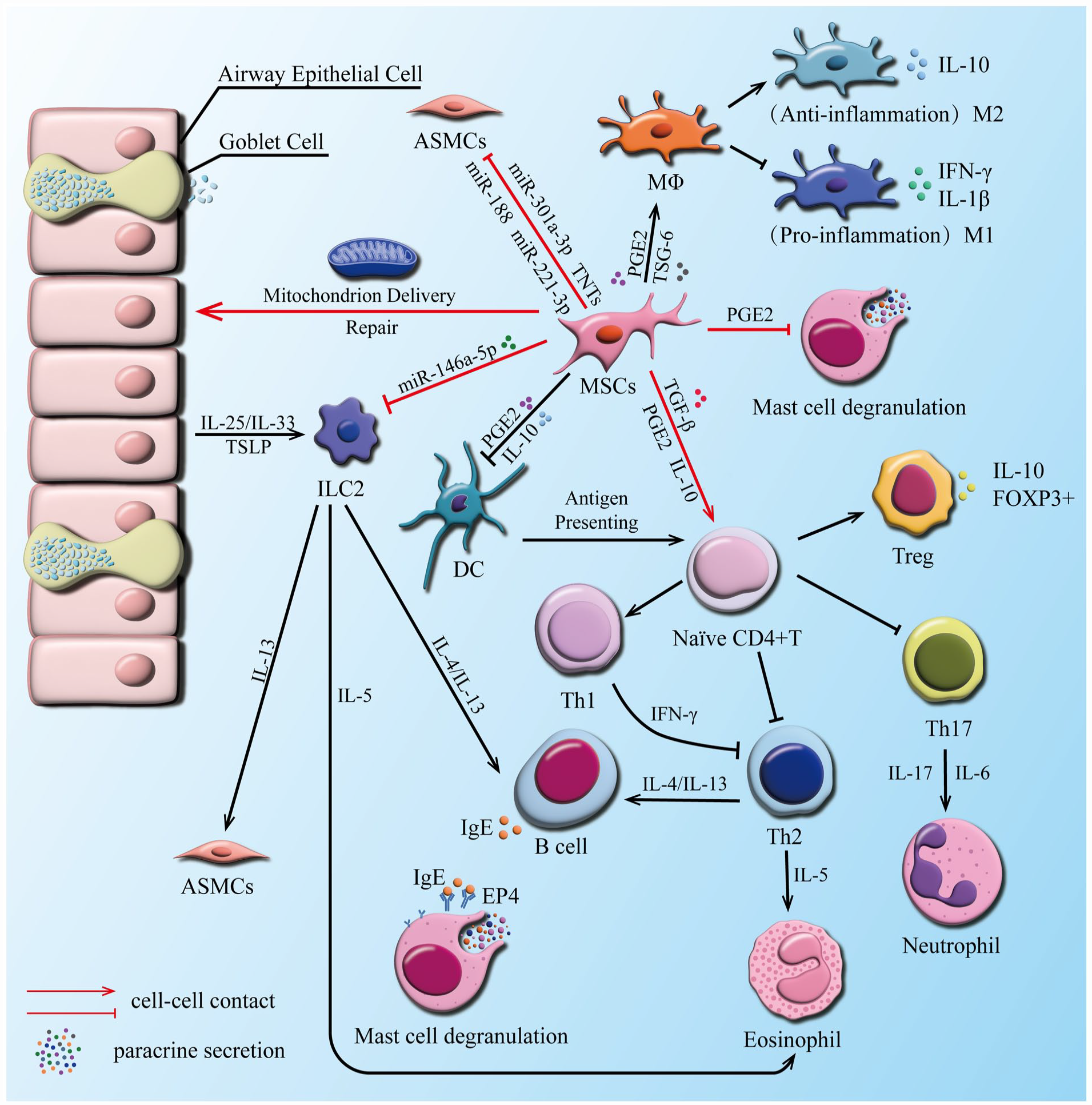
The crosstalk between MSCs and effector cells in asthma. MSCs reduce the production of various types of inflammatory factors (e.g., IL-4, IL-5, IL-13, IL-1β), promote the production and release of anti-inflammatory factors (e.g., IL-10, IDO, TGF-β, PGE2, TSG-6), and suppress the inflammatory response of ILC2, DCs, B cells, eosinophils, mast cells, and neutrophils. Moreover, MSCs restore Th1/Th2 balance, reverse Th17/Tregs imbalance, repair damaged epithelial cells, and promote the differentiation of anti-inflammatory M2 macrophages. MSCs: mesenchymal stem cells; TSLP: thymic stromal lymphopoietin; ILC2: innate lymphoid cell, group 2; ASMCs: airway smooth muscle cells; Th: helper T cell; DC: dendritic cells; Mø: macrophages; M1: M1 macrophages; M2: M2 macrophages; Treg: regulatory cells; PGE2: prostaglandin E2; TGF-β: transforming growth factor-β; IL: interleukin; Th: helper T lymphocyte; miR: microRNA; HO-1: heme oxygenase 1; TSG-6: tumor necrosis factor–stimulated gene 6; FOXP3+: forkhead box protein P3; IFN-γ: interferon (IFN)-γ; EP4: prostaglandin E2 receptor 4; IgE: immunoglobulin E.
Paracrine secretion
Several studies have shown that intratracheal or intravenous delivery of MSC-conditioned medium significantly reduces pathological injuries in rats sensitized to ovalbumin (OVA)64,82, indicating that paracrine secretion is a critical mechanism by which MSCs exert immunomodulatory effects in asthma. Paracrine secretion involves the secretion of soluble factors and extracellular vesicle (EV) production.
Soluble factors secretion
The secretion of soluble factors, such as TGF-β, prostaglandin E2 (PGE2), tumor necrosis factor–stimulated gene 6 (TSG-6), IL-10, indoleamine 2,3-dioxygenase (IDO), is critical for the immune-suppressive effects of MSCs 42 .
TGF-β is a critical regulator of Foxp3 expression that inhibits Th2 differentiation and promotes Treg production 42 . BM-MSC-derived TGF-β is essential for suppressing allergic responses in a ragweed-induced mouse asthma model 83 . The binding of IL-4, IL-13, or both in the allergic environment to IL-4R receptors on BM-MSCs activates the signal transducer and activator of the transcription 6 (STAT6) pathway, leading to increased TGF-β production, which activates TGF-β receptors in immune cells and leads to a decrease in IL-4 production, ultimately impelling immunological equilibrium. BM-MSCs derived from TGF-β1-knockout (KO) mice show no beneficial effects 83 . Paradoxically, studies have shown that TGF-β has adverse effects on asthma. Zhong et al. 15 demonstrated that TGF-β plays a pro-inflammatory role by showing that iPSC-MSCs attenuated airway inflammation in chronic allergic asthma by inhibiting the TGFβ1-Smad2/Smad3 signaling pathway. TGF-β1 is a key pro-fibrotic growth factor that contributes to the pathogenesis of airway remodeling in chronic asthma. It is responsible for myofibroblast extracellular matrix (ECM) synthesis and differentiation by inducing phosphorylation of its downstream targets Smad2 and Smad3 and stimulating α-smooth muscle actin84,85 The discrepancy in the role of TGF-β in vivo has not been fully elucidated, and the detailed mechanisms need to be further studied under different defined conditions.
PGE2 binds to E-prostaglandin 2 (EP2) and EP4 receptors expressed on the surface of immune cells and exerts pro-inflammatory effects. It induces the differentiation of DCs into an anti-inflammatory phenotype, suppresses the inflammatory response of MCs, and promotes the differentiation of Foxp3+Treg cells and the production of M2 macrophages42,79. Administration of mouse-AT-MSCs in a mouse asthma model elevates TGF-β and PGE2 in the lungs and improves lung function 86 . Blocking PGE2 and neutralizing TGF-β inhibits the production of Treg cells induced by mouse-AT-MSCs and eliminates the immunosuppressive effect of mouse-AT-MSCs in asthma 87 .
IL-10 inhibits the expression of co-stimulators, production of inflammatory factors, maturation of antigen-presenting cells, the proliferation of T cells, formation of memory T cells, and production of Th17 cells. Furthermore, it promotes Treg production 42 . Transplantation of human-PL-MSCs into asthmatic rats significantly increases IL-10, Foxp3, and Treg levels in lymph nodes, peripheral blood, and lung tissue. In addition, it decreases the levels of RAR-related orphan receptor γt (RORγt), a major transcription factor of IL-17 52 .
TSG-6 downregulates lymphocyte and neutrophil proliferation; decreases metalloproteinase activity and the expression of pro-inflammatory factors, IL-6, and IFN-γ; and promotes the expansion of Foxp3+Treg and M2 macrophages 39 . Cytokine-stimulated human MSCs secrete TGS-6 in the lungs, which attenuates lung inflammation and repairs injured lung tissue by weakening the migration capacity of innate immune cells (neutrophils and macrophages)88–90.
IDO is a vital secretory factor that mediates the immunomodulation of MSCs. However, it does not appear to play a crucial role in the inhibition of allergic airway inflammation. AT-MSCs derived from IDO-KO mice exert immunosuppressive effects on allergic airway inflammation. Moreover, there is no significant difference in the therapeutic effect of AT-MSCs between wild-type (WT) and IDO-KO asthmatic mice 91 .
Although various soluble factors have been reported to be responsible for suppressing airway inflammation in asthma, most reports fail to prove that these secreted factors are derived directly from MSCs as opposed to other immune cells. Moreover, the detailed mechanism of the involvement of these factors in asthma treatment remains unclear. Consequently, labeling soluble molecules derived from MSCs to trace their functional pathway is of great value in MSC-based therapies for asthma.
Production of EVs
MSC-derived extracellular vesicles (MSC-EVs) consist of lipid bilayers with vesicles containing multiple bioactive factors (e.g., mRNA, long non-coding RNAs, and miRNA) 92 , which are essential for intercellular communication. They are usually classified as exosomes, apoptotic bodies, and microvesicles93,94. However, current methods for isolating EVs generally yield a mixed population, and there is no consensus on specific subtype markers 95 . Exosome-specific markers typically consist of tetraspanins CD9, CD63, and CD81, while common markers for EVs consist of flotillin, heat-shock 70 kDa proteins, and major histocompatibility complex class I and class II 96 . The role of apoptotic bodies and microvesicles in asthma has yet to be elucidated, while numerous reports have suggested that exosomes play a crucial role in MSC-based therapy for asthma.
For instance, human UC-MSC-derived exosomes (MSC-Exos) abrogate inflammation in severe steroid-resistant asthma by reshaping macrophage polarization by inhibiting tumor necrosis factor receptor–associated factor 1 97 . Intranasal delivery of human placenta–derived MSC-Exos significantly increases IL-10-producing alveolar macrophages in the lungs and alleviates asthma symptoms in mice 98 .
Numerous studies have confirmed that the contents of MSC-EVs play a crucial role in ameliorating pathological changes in animal models of asthma. For example, exosome miR-301a-3p derived from rat-AT-MSCs suppressed the remodeling and inflammation of airway smooth muscle cells (ASMCs) stimulated by platelet-derived growth factor-BB by targeting signal transducer and activator of transcription 3 (STAT3), a transcription factor that controls inflammatory response and immunity 99 . Human BM-MSC-derived exosomal miR-188 inhibits ASMC proliferation induced by TGF-β1 by inhibiting the JARID2/Wnt/β-catenin axis, thus reducing lung collagen deposition in asthmatic mice. JARID2 is an mRNA target for miR-188 that activates the Wnt/β-catenin signaling pathway 100 . Rat-BM-MSC-derived miR-221-3p suppresses the expression of fibroblast growth factor 2 and the phosphorylation of extracellular signal–regulated kinase 1/2 signaling, thereby inhibiting the proliferation, migration, and ECM deposition of ASMC and alleviating OVA-induced pathological changes in asthmatic mice 101 . Human MSC-EV–derived miR-146a-5p can be taken up by ILC2 and then indirectly regulate Th2 cytokines, leading to the reduction of airway inflammation, mucus secretion, and elimination of AHR 102 . Human MSC-Exo–derived miR-1470 promotes CD4+CD25+FOXP3+ Treg differentiation in asthmatic patients by enhancing the expression of P27KIP1, an essential gene for CD4+CD25+FOXP3+ Treg differentiation 103 .
In addition, considering the positive role of EVs in asthma, Elga et al. artificially generated EV-mimetic nanovesicles (NV) from human-BM-MSCs, which have a yield higher than and a therapeutic potential comparable to that of natural EVs. MSC-derived NVs reduced cytokine production and eosinophil infiltration in bronchoalveolar lavage fluid (BALF) and lung tissues, thereby eliminating airway inflammation in an OVA-induced asthmatic mouse model 104 .
However, in addition to their contents, the function of MSC-EVs also depends on the expression of surface biomarkers. Cellular targeting, uptake, and plasma membrane fusion of EVs, which involves signaling membrane proteins, expression of cell surface receptors, and cell membrane fusion or endocytosis, are worth investigating.
Cell–cell contact
In a mouse model of asthma, human iPSC-MSCs–conditioned media injection reduced lung inflammation and increased lung function; however, the reduction was not as significant as that of transplanted MSCs 105 . This suggests that in addition to paracrine signaling, direct cell contact may be involved in the therapeutic role of MSCs in asthma. For instance, when iPSC-MSCs or BM-MSCs were co-cultured with PBMC from an allergic patient to allow cell-to-cell contact, iPSC-MSCs or BM-MSCs suppressed lymphocyte proliferation and the levels of inflammatory factors interferon-γ (IFN-γ), IL-4, IL-5, and IL-13, while enhancing IL-10 levels and Treg expansion in PBMCs 106 . In contrast, transwell separation eliminated the MSC-mediated immunosuppressive effect on PBMCs. Moreover, compared to the transwell system, co-cultured BM-MSCs exerted a better inhibitory influence on MC degranulation, chemokinesis, chemotaxis, and pro-inflammatory cytokine production 79 .
Cell–cell contact manifests through receptor attachment or channel formation. In iPSC-MSC–activated T-cell co-culture, the products of activated T cells stimulate the expression of adhesion molecules ICAM-1 and VCAM-1 in MSCs, making MSCs more adhesive to activated T cells and suppressing their proliferation. The opposite effects were achieved when ICAM-1 and VCAM-1 gene were knocked out or their expression was inhibited by blocking Abs 107 . In iPSC-MSC/ILC2s co-culture, iPSC-MSCs suppress ILC2 function by enhancing IL-10 secretion by Tregs through ICOS–ICOSL interaction, thereby lowering allergy-specific inflammation 80 . ICOS is expressed on Treg cells and ICOSL is expressed on iPSC-MSCs. Tunneling nanotubes (TNTs) serve as bridges between iPSC-MSCs and ASMCs or epithelial cells. Systemic administration of iPSC-MSCs rescued mitochondrial dysfunction and alleviated airway inflammation by delivering mitochondria from iPSC-MSCs to ASMCs and epithelial cells via TNTs in a mouse model of asthma19,108. Using the inflammatory chemotaxis capabilities of MSCs membranes to encapsulate normal mitochondria for the targeted delivery and repair of injured airway epithelial cells may be a potential therapeutic technique.
Strategies to Enhance the Therapeutic Effect of MSCs on Asthma
Although many preclinical trials have confirmed the positive effects of MSCs in asthma, there is still potential for advancement in the field. Pretreatment or combination of chemical reagents, in vivo microenvironment simulation, and gene expression modulation contribute to maintaining the stemness of MSCs, improving their proliferation ability and homing efficiency, as well as their anti-inflammatory and immunomodulatory properties, improving their efficacy in asthma (Fig. 2).

Strategies to enhance the therapeutic effects of MSCs on asthma. Strategies, such as pretreatment or combination therapy with chemical agents, mimicking the in vivo microenvironment, and genetic engineering, have been shown to boost MSC survival, maintain stemness, improve self-renewal, enhance homing efficiency, and improve anti-inflammatory and immunomodulatory capacity, thus inducing better remission of the pathogenesis of asthma. RLN: serelaxin; EPA: eicosapentaenoic acid; AA2G: ascorbyl 2-glucoside; CoCL2: cobalt chloride; Dex: dexamethasone; EPO: erythropoietin; sST2: soluble suppression of tumorigenicity 2; CX43: Connexin 43; IL-35: interleukin-35; IFN-γ: interferon-γ.
Mimicking the in vivo Microenvironment for MSC Pre-conditioning
Transplanted MSCs suffer from microenvironmental changes involving hypoxia (21% O2 in vitro culture, 2%–8% oxygen in the body) 29 , nutrient deficiency 109 , increased levels of oxidative stress 23 , and recruitment of immune cells post-transplantation 110 , all of which hamper the therapeutic potential of MSCs25,111,112. Culturing MSCs in media with hypoxia, inflammatory cytokines, or asthmatic serum to mimic the in vivo microenvironment can improve the survival or therapeutic potential of MSCs.
EVs secreted from hypoxia-pretreated human-UC-MSCs exhibit enhanced anti-inflammatory and anti-fibrotic potential by reducing the expression of pro-inflammatory factors, including IL-4 and IL-13, inhibiting the expression of α-smooth muscle actin and collagen-1, downregulating the TGF-β1-p-smad2/3 signaling pathway, and upregulating miR-146a-5p levels 29 . Moreover, hypoxia pretreatment suppresses eosinophil recruitment, inhibits ILC2 proliferation and function, reduces epithelial cell apoptosis, suppresses cupped cell proliferation, inhibits fibroblast proliferation and ECM deposition, decreases mucus production, reduces AHR, alleviates or reverses airway remodeling29,113. Serum from HDM-induced asthmatic mice stimulated with mouse-BM-MSCs suppress asthmatic airway inflammation and abrogated airway remodeling, as well as induce macrophage polarization to the anti-inflammatory M2 phenotype and increases the expression of anti-inflammatory mediators (e.g., TGF-β1, IFN-γ, IL-10, TSG-6, IDO-1, and IL1RA) and decreases fibroblast proliferation and ECM deposition induced by IL-4, IL-13, and eotaxin 27 . IFN-γ-pretreated dental follicle–derived MSCs increase the inhibitory effect of HDM-stimulated MSCs on lymphocyte apoptosis, promote the production of CD4+CD25+ FOXP3+Treg cells, and increase CD4+ T lymphocyte viability in asthmatic patients 114 . In an OVA-induced mouse model of asthma, IFN-γ-induced mouse-AT-MSCs significantly reduced pro-inflammatory factor IL-9 levels in serum and lung tissue, while increasing the expression of the anti-inflammatory factor FOXP3 in the lungs. Furthermore, IFN-γ-induced mouse-AT-MSCs decreased serum IL-9 levels and airway inflammation to a greater extent than non-induced MSCs 81 .
To facilitate the optimal and safe use of MSCs for basic research and therapeutic purposes, it is important to define optimal in vitro culture conditions for MSCs and to identify key mediators involved in MSC characterization.
Pretreatment or Combination Therapy With Chemical Agents
Pretreatment of MSCs with various chemical agents contributes to maintaining their stemness and enhancing their survival, anti-inflammatory, and immunomodulatory capacities, thus optimizing the regulatory role of MSCs in asthma.
Antioxidants: Ascorbic acid 2-glucoside (AA2G) is a vitamin C derivative with antioxidant effects. It maintains the stemness of MSCs and promotes their proliferation, mobilization, self-renewal, engraftment, homing, and anti-inflammatory properties 115 . In OVA/poly(I: C)-sensitized mice, AA2G-treated human-UC-MSCs reduced the infiltration of inflammatory cells, including macrophages, neutrophils, and lymphocytes, as well as the levels of pro-inflammatory mediators in BALF, including tumor necrosis factor α (TNF-α) and IL-17. Moreover, it increased the levels of IL-10, an anti-inflammatory cytokine, and the expression of TGS6, an anti-inflammatory gene 115 .
Anti-fibrotic drugs: Serelaxin (RLN) is an anti-fibrotic agent that enhances the viability and regenerative capacity of MSCs by creating a favorable environment for stem cell survival in diseased tissues. In OVA-induced chronic asthma, human-BM-MSCs combined with RLN effectively reduced AHR and reversed airway fibrosis owing to the synergistic effects of matrix synthesis inhibition by RLN and matrix metalloproteinase–mediated collagen degradation promotion by MSCs116,117.
Polyunsaturated fatty acids: Eicosapentaenoic acid (EPA) is an ω-3 polyunsaturated fatty acid with anti-inflammatory and immunomodulatory properties. In HDM-induced allergic asthma mice, EPA-pretreated mouse-BM-MSCs reduced the expression of pro-inflammatory mediators, such as IL-13, IL-4, and vascular endothelial growth factors, which enhanced the expression of anti-inflammatory factors, such as resolvin D1, PGE2, IL-10, and TGF-β, and reduced the total cell count in the BALF, BM, and lymph nodes. In addition, it regulated macrophage differentiation toward the anti-inflammatory M2 phenotype, relieving airway constriction, attenuating alveolar epithelial collapse, and reducing airway collagen fiber deposition 30 .
Low oxygen simulation compounds: Cobalt chloride (CoCl2), a hypoxia-mimetic compound, enhances the anti-inflammatory capacity of human-UC-MSCs through hypoxia-inducible factor 1 a (HIF-1a)-miR-146a-mediated signaling pathway, leading to a more significant reduction in lung infiltration of inflammatory cells, including macrophages, neutrophils, and lymphocytes in an OVA and poly(I: C)-induced mouse model of asthma 113 .
Statins: Simvastatin, a statin, reduces neutrophil and eosinophil infiltration, alleviates pulmonary inflammation and allergic responses in asthma, and improves lung compliance, but fails to reduce IL-13 and TGF-β levels 118 . Simvastatin combination facilitates the migration of mouse-BM-MSCs to the site of lung inflammation, reducing serum-specific IgE levels and lung levels of IL-13 and TGF-β, thereby decreasing goblet cell hyperplasia and collagen deposition119,120.
Corticosteroid: Dexamethasone (Dex) in combination with induced pluripotent stem cells and mesenchymoangioblast-derived mesenchymal stem cells (MCA-MSCs) reduced goblet cell metaplasia and goblet cell count to a significantly greater extent than MCA-MSCs alone 20 .
Ferroptosis inhibitor: Liproxstatin-1 is an inhibitor of ferroptosis. Compared with naïve MSCs in chronic asthma mouse models, liproxstatin-1-primed human-UC-MSCs showed better downregulation of Th2 cell activation, Ly6C + M2 macrophage population, airway inflammation, and fibrosis 50 . Ly6C is a marker of pro-inflammatory and pro-fibrotic phenotypes 57 .
Pretreatment or loading of MSCs with small-molecule drugs or biologics to increase surface receptor expression or immunosuppressive factor secretion may be a promising strategy to improve MSCs’ efficacy.
Genetic Engineering
Modulating the expression of target genes in MSCs using genetic engineering is a promising way to monitor the duration of efficacy of transplanted MSCs or improve their therapeutic effectiveness in asthma.
For instance, the green fluorescent protein was transfected into mouse-AT-MSCs to track cell fate after intravenous administration to test the efficacy of MSCs in animals with persistent allergic asthma 60 . Connexin 43 (CX43) mediates TNT formation between epithelial cells and iPSC-MSCs. Human-iPSC-MSCs deliver mitochondria to epithelial cells via TNT to protect the latter cells from injury and apoptosis induced by mitochondrial dysfunction. In an OVA-induced asthma mouse model, CX43 overexpression in iPSC-MSCs (MSCs-CX43) enhances the formation of TNT between epithelial cells and iPSC-MSCs as compared to iPSC-MSCs, thereby exhibiting better performance in alleviating airway inflammation and mucus secretion 19 . IL-35 has a regulatory effect on CD4+ T-cell-mediated immunity and Th17 response 121 . In an OVA-sensitized asthmatic mouse model, MSCs transfected with the gene encoding IL-35 exerted a more substantial effect on controlling allergic asthma symptoms than MSCs without IL-35 122 . Erythropoietin (EPO) has antioxidant and anti-inflammatory properties that enhance MSC proliferation and migration. In a mouse model of OVA-induced asthma, EPO-MSCs inhibited airway inflammation, mucus secretion, AHR, and airway remodeling by downregulating TGF-β1-TAK1-p38MAPK pathway activity 26 . sST2 is a decoy receptor for IL-33, which can initiate Th2-associated cytokine production. In occupational asthma induced by ammonium persulfate, human-AT-MSCs overexpressing soluble suppression of tumorigenicity 2 prevented IL-33 production, decreased IgE levels, and alleviated neutrophilic inflammation and airway remodeling 123 .
In addition, human-MSCs modified with the miR-138-5p inhibitor exerted a greater capacity to attenuate inflammation and allergic responses by activating the expression of sirtuin 1 (SIRT1) and inhibiting the HMGB1/TLR4 pathway in an OVA-induced asthma mouse model 28 .
Despite improved therapeutic effects observed in various asthma models, the use of viral vectors remains a safety concern. Therefore, the long-term safety of genetically modified MSCs should be investigated in preclinical models
Clinical Trials
Although reports regarding MSC-based therapy for patients with asthma are rare, a growing number of clinical trials involving the use of MSCs for asthma are being registered on ClinicalTrials.gov (https://clinicaltrials.gov) as MSCs have been proven to have positive effects on animal models of asthma. As of December 2022, approximately four clinical trials of MSCs treatment in patients with asthma have been registered.
In a phase II clinical trial performed at Punta Pacifica Hospital of Panama City (NCT02192736, July 17, 2014), intranasal administration of mesenchymal trophic factors (MTF) derived from allogenic human-UC-MSCs once daily for 4 weeks to assess the safety of MTF and its effect on airflow limitation in asthma patients. In a phase I clinical trial at Medical Surgical Associates Center St. John’s, Antigua, and Barbuda (NCT05147688, December 7, 2021), human-UC-MSCs were administered via a single intravenous infusion (1 × 109) in patients with lung diseases (including asthma). Their safety and effect on lung function were assessed within 1 month before treatment and at 1, 6, 12, 24, 36, and 48 months after human-UC-MSCs treatment. Another phase I clinical trial (NCT03137199, May 2, 2017) at the University of Miami Miller School of Medicine investigated the safety, tolerance, and effect of allogeneic human-BM-MSCs on lung function and airway inflammation in asthma patients through peripheral intravenous infusions (2 × 107 and 1 × 108) for 48 weeks. In another phase I clinical trial at Children’s Healthcare of Atlanta (NCT05035862, September 5, 2021), human-UC-MSCs and allogeneic IFN-γ-primed human-BM-MSCs were administered via a single intravenous infusion in patients with moderate to severe asthma at a dose of 2 × 106 cells/kg or 5 × 106 cells/kg. Seven to 30 days after administration, the safety of MSCs treatment was evaluated by assessing side effects, including dyspnea, coughing, wheezing, respiratory failure, allergic reactions, and infusion-related reactions. The improvement of lung function and airway inflammation in these patients was also monitored (Table 2).
Clinical Trials Concerning MSC-Based Therapy on Asthma Registered on ClinicalTrials.gov.
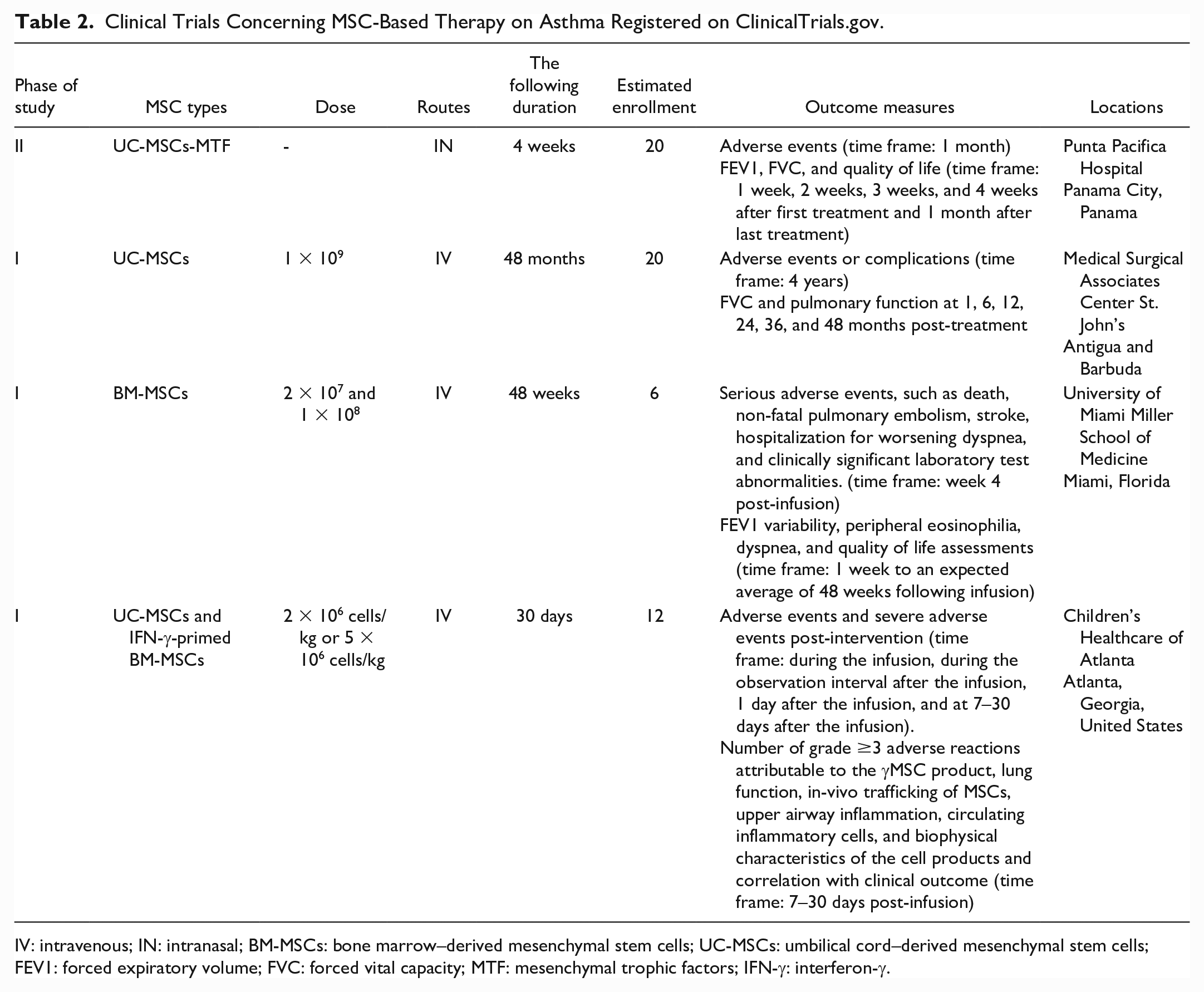
IV: intravenous; IN: intranasal; BM-MSCs: bone marrow–derived mesenchymal stem cells; UC-MSCs: umbilical cord–derived mesenchymal stem cells; FEV1: forced expiratory volume; FVC: forced vital capacity; MTF: mesenchymal trophic factors; IFN-γ: interferon-γ.
In addition, a 68-year-old man with persistent asthma symptoms who enrolled in a phase 1 clinical trial received an intravenous injection of 100 million human-UC-MSCs. Then, 2- and 6-month post-treatment follow-ups were performed. During several months of follow-up, no treatment-related adverse events or complications were observed. Within 2 months of treatment, the frequency of rescue inhaler use was gradually reduced to once per month, with a reduction of more than 90%. Moreover, his nebulizer use was reduced by 70% 124 . Unfortunately, despite positive results reported in the case report, the clinical trial has not yet given a definitive conclusion on the safety and efficiency of MSCs in the treatment of asthma.
Conclusion and Future Perspective
MSCs are characterized by easy accessibility, only a few ethical concerns, multipotent differentiation capacity, and immunomodulatory properties, all of which make them promising therapeutic candidates for asthma. Although there have been many reports on the use of MSCs in the treatment of asthma over the past few years, no clinical trials have been reported. Before MSCs can be safely, effectively, and routinely applied in clinical settings, the following issues remain to be resolved.
MSC heterogeneity: Different MSC types exert distinct characteristics and secretion profiles, which may result in distinct therapeutic effects in asthma. A better understanding of the optimal sources of MSCs and their underlying mechanisms may help maximize the therapeutic effect of MSCs.
Consensus on MSC therapy: The efficacy of MSCs for asthma varies depending on the routes, dose, and timing of MSCs. Nowadays, there is a lack of consensus on MSC-based therapy for asthma.
MSC senescence: Human MSC–based cell therapy requires tens of millions of cells, but the number of cells initially isolated from adult tissues is low. Therefore, MSCs must be cultured for a long time to reach the cell volumes required for therapy. However, during long-term cultivation, MSCs gradually lose their expansion capacity and stemness, leading to a reduction in therapeutic potential, which may subsequently adversely affect outcomes after transplantation. Consequently, rejuvenating MSCs is a powerful strategy for improving their therapeutic potential. For example, SIRT1 is a classical antisenescence gene associated with asthma 125 . SIRT1-modified MSCs can delay senescence and alleviate airway inflammation in asthma. In our ongoing study on asthma treatment, we over-expressed SIRT1 in BMSCs via lentivirus and found that SIRT1 can significantly attenuate oxidative stress-induced BMSCs senescence, as evidenced by reduced SA-β-gal positive cell ratio and lowered the levels of senescence-related proteins (p16, p21, and p53). Furthermore, compared to wild-type BMSCs, SIRT1-modified BMSCs significantly reduced the number of inflammatory cells in BALF and lowered lung inflammation scores when injected into asthma model mice via tail vein (Our unpublished data).
Safety issues: First, MSC therapy poses quality challenges. The inconsistent standardization in the preparation process and variations in donor age, gender, physical condition, cell culture, storage, transportation, recovery, and preparation can all contribute to the uneven quality and low survival rates including cell death of MSCs. Moreover, various sources of infection may be introduced during transplantation such as viruses like HIV or HBV or fungi that can lead to adverse reactions. Second, MSC therapy faces the risk of immune rejection. Despite their low immunogenicity and immunomodulatory effects, MSCs cannot be deemed immune privileged. For instance, infusion of allogeneic MSCs induces immune memory while also stimulating innate immune responses126,127. Furthermore, if the immunosuppressive program of MSCs is not activated, they can function similarly to antigen-presenting cells and foster inflammation in vivo 128 . Transplanted MSCs that have been exposed to IFN-γ or differentiated into mature cells exhibit markedly increased expression of MHC class I and MHC class II molecules129,130. Adverse effects such as fever, chills, headache, back pain, and numbness have been observed after MSC transplantation43,131. Recent studies have revealed that delivering human AT-MSCs intranasally to non-inflamed mouse lungs resulted in a low-grade lung inflammation with increased IFN-γ expression, weakened epithelial barrier integrity due to decreased tight junction proteins, activation of innate and adaptive immune responses with elevated IL-7 and T/B cell signaling pathways, and induced redox imbalance characterized by increased HIF1a signaling and superoxide radical degradation. These effects could be attributed to the activation of the recipient organ’s hypoxia pathways in vivo by the transplanted MSCs, which resulted in MSC apoptosis and then triggered a series of graft clearance reactions 132 . Third, the uncontrollable differentiation of MSCs during transplantation has become a safety hazard associated with its application. Last, MSCs have tumor-tropic and tumor-promoting properties, which is of concern to researchers. Previous studies have shown that prolonged proliferation of MSCs in vitro induces cytogenetic abnormalities and subsequent differentiation into tumor cells after in vivo administration 133 . MSCs interact with carcinoma cells to promote cancer progression and drug resistance 134 .
Collectively, the understanding of MSCs in treating asthma is still limited for the limited reports. There is a need to explore their targets and mechanisms in asthma comprehensively and track their prognosis in vivo including long-term implantation consequences and adverse reactions after transplantation. Standardized administrations should also be established to ensure the safe and effective utilization of MSC therapy.
Footnotes
Author Contributions
JZ, YC, NC, XZ, DZ, and RC reviewed the references and collected data. SH and YL edited the manuscript and supervised the study. GM and YW critically revised the manuscript. All authors read and approved the current version of the manuscript.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support for this work includes funding from the National Natural Science Foundation of China (81770034 and 81670252); Guangdong Basic and Applied Basic Foundation (2019A1515011306, 2020A1515010240, 2022A1515140167, and 2022A1515110234); State Administration of Traditional Chinese Medicine of Guangdong Province, China (20231319); Doctoral Scientific Initiated Project of Shunde Women and Children’s Hospital of Guangdong Medical University (2020BSQD003 and 2021BSQD001); 2022 Competitive Support Talent Project of Shunde District (No. 8).