
Abstract
Endometrial cancer is the most common gynecologic cancer with high heterogeneity. However, there are limited treatment options for advanced endometrial carcinoma. In recent years, immunotherapy has broadly been used for the treatment of various cancers. However, the efficacy of immunotherapy against endometrial cancer is limited. The tumor microenvironment, including mesenchymal stem cells (MSCs), may contribute to tumor progression through cancer cells themselves and through cells of the immune system. We successfully isolated endometrial cancer–derived MSCs (EmCaMSCs) from patients and found that the population of MSCs in tumor tissues was approximately 1%–5%. The population of MSCs correlated with the stage of the disease. EmCaMSCs expressed MSC markers and exhibited trilineage differentiation ability. The programmed death ligands PD-L1 and PD-L2 were highly expressed in EmCaMSCs; their expression could be further enhanced by tumor necrosis factor-α and interferon-γ. When cocultured with peripheral blood mononuclear cells (PBMCs), anti-CD3, and anti-CD28, EmCaMSCs inhibited the proliferation of PBMCs, which were partially rescued by treatment with anti-PD-L1 antibodies. From the profile of conditioned medium of EmCaMSCs, we discovered that interleukin (IL)-8 and insulin-like growth factor–binding protein 6 could also rescue the proliferation of PBMCs. Furthermore, EmCaMSCs cocultured with IL-2-induced PBMCs exhibited decreased expression of CD56, CD4, and CD8 and showed decreased cytotoxicity toward K562 cells and endometrial cancer cells. Overall, EmCaMSCs were isolated successfully from endometrial cancer tissues and exhibited immunosuppressive effects and may be targeted for the treatment of endometrial cancer by anti-cytokine and immune checkpoint inhibitors. The percentage of MSCs in tumor stroma might predict the prognosis of endometrial cancer.
Introduction
Uterine cancer is the most common gynecologic cancer in the United States and Taiwan1,2, and endometrial cancers comprise more than 90% of all uterine cancers 2 . Unopposed estrogen secretion contributes to the risk of endometrial cancer. Current treatments are staging surgery and adjuvant radiotherapy or chemotherapy for advanced stage 3 . However, women with recurrent or metastatic endometrial cancer have limited treatment options and show poor prognoses. Therefore, there is an urgent need for new therapies for the treatment of this disease 4 .
Endometrial cancer can be divided into four molecular subtypes including the polymerase ε (POLE) ultra-mutated and microsatellite instability (MSI) hypermutated types 5 . POLE ultra-mutated and MSI-high endometrial cancers are abundantly infiltrated with T cells and show high expressions of high programmed death-1 (PD-1) and programmed death-ligand 1 (PD-L1) 6 . Inhibitors of PD-1 have been developed as potential approaches for the treatment of endometrial cancers 7 .
Tumor microenvironments may harbor immunosuppressive agents, such as cancer-associated mesenchymal stem cells (CaMSCs) 8 . CaMSCs can secrete various cytokines to promote tumor progression. CaMSC-secreted interleukin (IL)-6 can enhance chemoresistance and tumor progression9,10. CaMSC-secreted IL-8 is involved in promoting tumor progression 11 . Above all, CaMSCs may promote tumor progression via specific cytokines.
We hypothesized that endometrial CaMSCs may exhibit cytokines and PD-L1-mediated immunosuppressive effects in the tumor microenvironment. This study aimed to analyze whether endometrial CaMSCs harbor PD-L1-mediated immunosuppression or cytokines to promote cancer in the tumor microenvironment.
Materials and Methods
Ethics
This study was approved by the Research Ethics Committee of Hualien Tzu Chi Hospital (IRB 109-257-A), and informed consent was obtained from all the patients included in the study. We confirm that all experiments were performed in accordance with the relevant named guidelines and regulations.
Endometrial cancer patients were included if they were >18 years of age, female, and had been admitted for staging surgery after biopsy-proven endometrial cancer.
Clinical Specimens and Isolation of Endometrial CaMSCs
The specimens of endometrial cancer tissue were cut into small pieces, digested with type I collagenase (Worthington Biochemical Corporation, Lakewood, NJ, USA), and incubated for 1–2 h at 37°C in a humidified atmosphere under 5% CO2. The samples were passed through 70 μm and 40 μm cell strainers (Falcon) to collect single cells. After washing out the collagenase with phosphate-buffered saline (PBS), the cells were seeded into six-well plates containing low-glucose Dulbecco’s Modified Eagle medium (DMEM; Gibco, Waltham, MA, USA), supplemented with 10% fetal bovine serum (FBS; Gibco) and 1× penicillin/streptomycin (Gibco). The medium was refreshed, and cells were expanded every 2–3 days. Briefly, endometrial cancer–derived MSCs (EmCaMSCs) before P8 (8-passages) were used in the experiments.
Isolation of Bone Marrow MSCs and Wharton’s Jelly MSCs
Bone marrow MSCs (BMSCs) were collected under ethical approval (IRB number: IRB103-49-A). Bone marrow was aspirated from patients with bone trauma who underwent orthopedic surgery. First, 5 ml of bone marrow aspirate was mixed with 5 ml of PBS. Then, the mixed bone marrow aspirate was centrifuged at 1,200 × g for 6 min, and the supernatant was removed. The resultant pellet was plated on a culture dish with a culture medium (alpha-MEM, 15% FBS, and 1% penicillin/streptomycin).
All the experiments that required collection of Wharton’s jelly MSCs (WJ-MSCs) were approved by the Research Ethics Committee of Buddhist Tzu Chi General Hospital (IRB 100-166), and written consent was obtained from all study participants. The detailed derivation protocol for WJ-MSCs has been previously reported 12 . Briefly, one human umbilical cord sample (20 cm in length, 20 g in weight) was collected, and separation of Wharton’s jelly from the vessels and the amniotic membrane was conducted within 24 h. The jelly was then cut into pieces smaller than 0.5 cm3, treated with collagenase type 1 (Sigma, St Louis, MO, USA), and incubated for 14–18 h at 37°C in a 95% air/5% CO2 humidified atmosphere. The explants were then cultured in low-glucose DMEM containing 10% FBS and antibiotics at 37°C in a 95% air/5% CO2 humidified atmosphere. They were left undisturbed for 5–7 days to allow for the migration of cells from the explants. The resultant MSCs were WJ-MSCs.
Flow Cytometry
The cells were stained with fluorescent dye–conjugated antibodies for 30 min on ice and washed twice with PBS. After resuspending in PBS containing 2% FBS, the cells were analyzed by fluorescence-activated cell sorting (FACS Calibur; BD Biosciences, Franklin, NJ, USA). The following antibodies were used in the analysis: CD34-FITC, CD73-FITC, CD90-FITC, CD90-PE, human leukocyte antigen (HLA)-ABC-FITC, HLA-DR-FITC (eBioscience, San Diego, CA, USA); CD44-FITC, CD105-APC (BD Biosciences); CD45-FITC (BioLegend, San Diego, CA, USA). In the case of fresh tissues, CD73-FITC, CD90-PE, and CD105-APC were used for triple staining. Antibodies against PD-L1-PE and PD-L2-APC were purchased from eBioscience.
Differentiation Into Adipocytes, Osteoblasts, and Chondrocytes
For adipogenesis, EmCaMSCs were cultured in DMEM containing 10% FBS, 1 mmol/l dexamethasone, 5 mg/ml insulin, 0.5 mmol/l isobutylmethylxanthine, and 60 mmol/l indomethacin (all from Sigma-Aldrich, St. Louis, MO, USA) for 2 weeks. Cells were stained with Oil Red O (Sigma-Aldrich); the intracellular lipid droplets showed positive signals. To investigate osteogenesis, EmCaMSCs were cultured in DMEM supplemented with 10% FBS, 0.1 mmol/l dexamethasone, 10 mmol/l β-glycerol phosphate, and 50 mmol/l ascorbate for 2 weeks. Positive staining with alizarin red S (Sigma-Aldrich) was indicative of mineralized calcium deposits, which are characteristic of osteoblasts. For chondrogenesis, EmCaMSCs were loaded into the bottom of 15 ml conical tubes containing DMEM medium supplemented with 10% FBS, 10 ng/mL transforming growth factor β1 (PeproTech, Rocky Hill, NJ, USA), 50 μg/ml ascorbic acid 2-phosphate (Sigma-Aldrich), and 6.25 μg/ml insulin (Sigma-Aldrich) and incubated for 3 weeks. The formed cartilage was fixed in 4% paraformaldehyde for 24 h before transferring to 70% ethanol for an additional 24 h. It was then embedded in an optimal cutting temperature compound, cryosectioned, and stained with Safranin O, Aggrecan, and type II collagen.
Immunohistochemical Analysis
Briefly, the cryosectioned samples of chondrocytes (5–8 μm) were stained with Safranin O, anti-Aggrecan, and anti–type II collagen (1:200; Sigma-Aldrich) at 4°C overnight. After washing with PBS/Tween, the sections were incubated with secondary antibodies at room temperature for 1 h, and further stained with 3,3′-diaminobenzidine chromogen and hematoxylin (Sigma-Aldrich). Brown coloration was an indicator of protein expression.
Quantitative RT-PCR
Total RNA was purified using a PureLink RNA Mini kit (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s instructions. Briefly, 1 μg of total RNA was reverse transcribed into cDNA using SuperScript III enzyme (Invitrogen, Carlsbad, CA, USA), as per the manufacturer’s instruction. A real-time polymerase chain reaction (RT-PCR) was performed using the Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and analyzed using Quant Studio 5 (Applied Biosystems). The glyceraldehyde-3-phosphate dehydrogenase gene was used as an internal control, and all of the experiments were conducted in triplicate. The primer sequences used are listed in Supplementary Table 1.
Induction of PD-L1 and PD-L2 Expression
EmCaMSCs were treated with tumor necrosis factor-α (TNFα) (10 ng/ml; Abcam, Cambridge, UK) and interferon-γ (IFNγ) (100 ng/ml; Abcam), or IFNγ alone for 3 days. The expression of PD-L1 and PD-L2 was detected using FACS. Co-staining was carried out using PE-anti-PD-L1 (eBioscience) and APC-anti-PD-L2 (eBioscience), and protein expression was analyzed in terms of the mean fluorescent intensity using the CellQuest software (Calibur; BD Biosciences).
PBMCs Cocultured With EmCaMSCs
EmCaMSC2 and EmCaMSC3 were seeded at a cell density of 1 × 105. After attaching CaMSCs, PBMCs were added and the cells were cocultured at different ratios (1:1, 1:2, 1:4, and 1:8), with or without IFNγ (100 ng/ml) treatment for 3 days. The cell viability was measured by using 2,3-bis-(2-methoxy-4-nitro-5-sulphenyl)(2H)-tetrazolium-5-carboxanilide (Biological Industries Ltd., Beit-Haemek, Israel). Briefly, the optical density was measured at 450 nm, and the absorbance at the reference wavelength of 650 nm was subtracted and adjusted with that of the blank control (wells without cells). All of the wells contained anti-CD3 (5 µg/ml) and anti-CD28 (5 µg/ml) antibodies to promote T-cell proliferation, and all experiments were conducted in triplicate. PBMC cells alone (0:1, 0:2, 0:4, and 0:8) were used as the positive control. For blocking or activating experiments, nivolumab (3,000 ng/ml; Opdivo), anti-PD-L1 (3,000 ng/ml; Abcam), recombinant IL-8 (300 ng/ml; ProSpect), anti-IL-8 (3,000 ng/ml; Abcam), anti-IGFBP2 (3,000 ng/ml; Santa Cruz Biotechnology, Dallas, TX, USA), and anti-insulin-like growth factor–binding protein 6 (IGFBP6) (3,000 ng/ml; Santa Cruz Biotechnology) were used.
Cytokine Array
First, 1 × 106 EmCaMSC2 or EmCaMSC3 cells were cultured in a 10-cm dish for 2 days, and the conditioned medium (CM) was collected and passed through a 0.22-μm cell strainer to remove cell debris. The cytokines from endometrial cancer–derived mesenchymal stem cell–conditioned medium (EmCaMSC-CM) were measured using a cytokine array kit (ab193656; Abcam) according to the instructions of the manufacturer. Each sample was divided into two parts for membrane C6 and C7, and a total of 120 cytokines were analyzed.
Enzyme-Linked Immunosorbent Assay (ELISA)
Concentrations of IL-8, IGFBP2, and IGFBP6 were determined using an ELISA Kit (Abcam) according to the instructions of the manufacturer. Briefly, the diluted CM of EmCaMSCs was incubated with antibody-coating plates for 1 h. After washing, the plates were incubated with a streptavidin-horseradish peroxidase solution for 30 min and chromogen for 10 min. The reaction was stopped and the absorbance measured at 450 nm for the primary signal and 620 nm for a reference signal. The results were calculated by OD450 minus OD620 and blank and fitted into the standard curve to determine the concentration of target proteins.
Cytotoxicity Assay of PBMCs Cocultured With EmCaMSCs
Briefly, PBMCs were cocultured with EmCaMSC2 and EmCaMSC3 in the presence of IL-2 (3,000 U/ml; Gibco) for 4 days. The ratio of PBMCs to EmCaMSCs was 10:1. PBMCs were harvested and then further cocultured with K562 cells in ratios of 5:1 and 10:1 for 4 h. The medium was replaced with PBS containing 2% FBS and fresh IL-2. Cells were harvested and stained with propidium iodide (PI) (Sigma-Aldrich) for 10 min. The K562 cells could be separated by forward scatter/side scatter, and the proportion of PI-positive K562 cells was calculated. For the cytotoxicity test in endometrial cancer cells, RL95-2 and HEC-1A were seeded and cultured overnight. After attaching cells, PBMCs were cocultured with EmCaMSCs as above or with anti-CD3/CD28 for 4 days and were further cocultured with cancer cells at a 10:1 ratio for 6 h with IL-2 or anti-IL-8, anti-IGFBP2, and anti-IGFBP6. The supernatants were collected for lactate dehydrogenase (LDH) activity test (Thermo-Fisher, Waltham, MA, USA) following the manufacturer’s instructions. PBMCs cultured alone and treated with IL-2 or anti-CD3/CD28 served as the control. All of the experiments were conducted in triplicate.
Statistical Analyses
We compared the two groups using an independent-samples t-test or Wilcoxon rank-sum test for continuous variables and the chi-square test for categorical variables. For more than two groups, we used analysis of variance (ANOVA) with a post hoc test (Bonferroni test). SPSS software (version 24; IBM, NY, USA) was used for statistical analysis. A P value <0.05 was considered statistically significant.
Results
Composition, Isolation, and Characterization of EmCaMSCs
Typical MSCs tend to express major MSC surface markers, including CD73, CD90, and CD105, and exhibit the ability to differentiate into adipocytes, chondrocytes, and osteoblasts 13 . Therefore, we used triple staining to determine the size of the MSC population (Fig. 1A). The proportion of EmCaMSCs was associated with the stage of the disease (P < 0.05, Fig. 1B).

The population of mesenchymal stem cells (MSCs) in endometrial cancer tissue. (A) Fresh endometrial cancer tissue was digested by type I collagenase into single cells. Cells were triple stained with CD73, CD90, and CD105; triple-positive staining was indicative of MSCs. The MSC population was calculated by multiplying the CD73+ population by the CD90+ CD105 double-positive population. (B) The percentage of MSCs between early-stage (stage I, n = 13) and advanced-stage (stages II and III, n = 3) endometrial cancer was noted. *P < 0.05.
Subsequently, the CaMSCs were successfully isolated from endometrial cancer patients and their nature was confirmed by analyzing other markers. Like MSCs obtained from other sources, EmCaMSCs expressed CD44, CD73, CD90, CD105, and HLA-ABC, but lacked CD34, CD45, and HLA-DR (Fig. 2A). Furthermore, these EmCaMSCs showed the ability to successfully differentiate into adipocytes (positive oil red staining), osteoblasts (positive alizarin red S staining) (Fig. 2B), and chondrocytes (positive Safranin O staining) (Fig. 2C). Immunohistochemistry also showed positive staining for aggrecan and type II collagen (chondrocyte markers) (Fig. 2C).

Characterization and trilineage differentiation of endometrial cancer–derived mesenchymal stem cells (EmCaMSCs). (A) Representative flow cytometry histograms of EmCaMSCs at passage 3. The EmCaMSCs were positive for CD29, CD44, CD90, CD105, and HLA-ABC, but negative for CD34, CD45, and HLA-DR. (B) After culturing in adipogenic media for 14 days, positive staining with Oil Red O was observed (upper panel). After culturing in osteogenic media for 14 days, osteogenesis was confirmed by positive staining with alizarin red (lower panel). The right panel shows the quantification of Oil Red and alizarin red OD ratio in differentiated adipocytes and osteoblasts. **P < 0.01, ***P < 0.001. (C) After 21 days of chondrogenesis, the formatted three-dimensional pallet showed positive Safranin O, aggrecan, and type 2 collagen staining. Scale bar = 100 µm. All of the EmCaMSCs for the experiments were conducted in triplicate.
Clinical Information of the Patients and Cell Characteristics of EmCaMSCs
The clinical information of the patients who provided EmCaMSCs is listed in Table 1. In the following experiments, we used EmCaMSC2 and EmCaMSC3 because there were no MSCs in the tumor for EmCaMSC1. One early-stage and one advanced-stage EmCaMSC were used for further experiments. The three patients were without classical risk factors such as hypertension, diabetes mellitus, and high body mass index (BMI). Two patients had childbirth histories. All three EmCaMSCs met classical MSC characteristics (CD marker panels and differentiation capabilities) 14 .
Clinical and Cell Characteristics of EmCaMSC Patients.
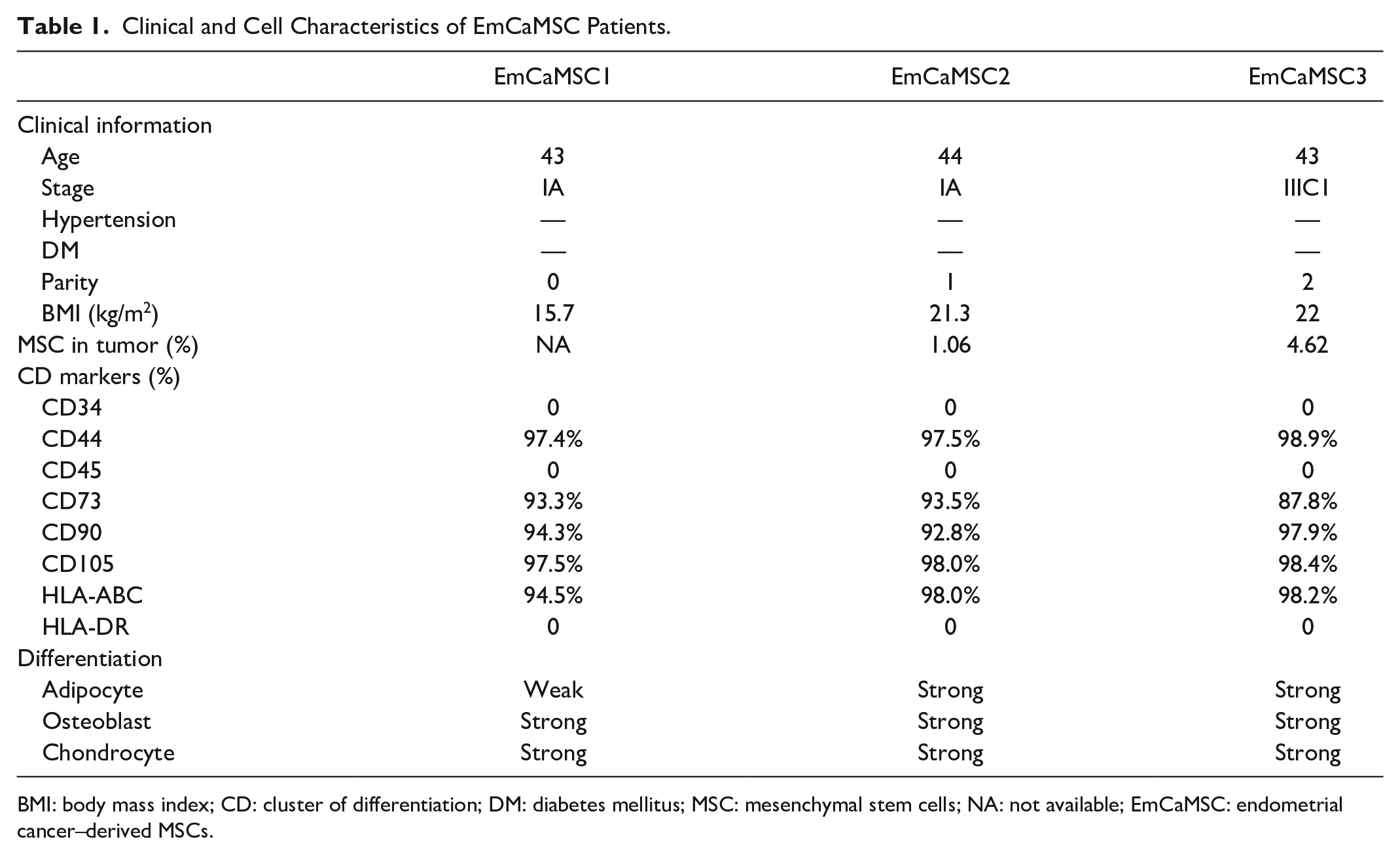
BMI: body mass index; CD: cluster of differentiation; DM: diabetes mellitus; MSC: mesenchymal stem cells; NA: not available; EmCaMSC: endometrial cancer–derived MSCs.
PD-L1 and PD-L2 Are Expressed in EmCaMSCs
Subsequently, we used quantitative RT-PCR (qRT-PCR) and flow cytometry to check whether EmCaMSCs expressed PD-L1 and PD-L2. qRT-PCR revealed that the PD-L1 and PD-L2 genes were expressed in EmCaMSCs (n = 3, Fig. 3A). Moreover, flow cytometry showed that protein expression could also be increased (~1.6-fold in PD-L1 and ~2.4-fold in PD-L2) by treatment with TNFα and IFNγ (Fig. 3B and C). Overall, gene and protein expressions of PD-L1 and PD-L2 were observed in EmCaMSCs.

Baseline and induced expression of programmed death ligands, PD-L1 and PD-L2, in endometrial cancer–derived mesenchymal stem cells (EmCaMSCs). The expression of PD-L1 and PD-L2 was analyzed by (A) quantitative reverse transcription-polymerase chain reaction and (B) fluorescence-activated cell sorting. After treating with tumor necrosis factor-α (10 ng/ml) and interferon-γ (100 ng/ml) for 24 h, the protein expression of PD-L1 and PD-L2 was (B) analyzed by fluorescence-activated cell sorting and (C) quantified on the basis of mean fluorescence intensity. All of the experiments were conducted in triplicate. ***P < 0.001.
EmCaMSCs Suppressed PBMC Proliferation and Could be Partially Reversed by Inhibiting PD-L1
Next, we evaluated the ability of EmCaMSCs to inhibit PBMC proliferation. We directly cocultured EmCaMSCs and PBMCs and treated them with anti-CD3 and anti-CD28 antibodies for 3 days to enhance T-cell proliferation. Figure 4A shows the morphology of EmCaMSCs (a), PBMCs (b), cocultured EmCaMSCs and PBMCs (c), and cocultured EmCaMSCs and PBMCs plus IFNγ (d). We noted that PBMCs formed large colonies when they were not cocultured; coculturing with EmCaMSCs reduced the size of the colonies. We found that the inhibition of PBMC proliferation was dependent on the ratio between EmCaMSCs and PBMC (1:1, 1:2, 1:4, and 1:8; P < 0.001; Fig. 4B). Treatment of EmCaMSCs with IFNγ significantly enhanced this inhibitory effect (Fig. 4C). After being treated with nivolumab and/or anti-PD-L1 antibodies, the proliferation of PBMCs (cocultured with EmCaMSCs) was increased 2- to 2.5-fold (P < 0.05) (Fig. 4D). These results demonstrated that EmCaMSCs could suppress the proliferation of PBMCs through cell–cell contact and could be blocked by inhibiting PD-L1.

The function of programmed death ligands, PD-L1 and PD-L2, in the proliferation of peripheral blood cells (PBMCs) cocultured with endometrial cancer–derived mesenchymal stem cells (EmCaMSCs). (A) The morphology of PBMCs cocultured with EmCaMSCs, anti-CD3 antibodies (5 µg/ml), and anti-CD28 antibodies (5 µg/ml) for 3 days. From left: EmCaMSCs only, PBMC only, PBMC cocultured with EmCaMSCs, PBMC cocultured with EmCaMSCs and interferon-γ. (B) The proliferation of PBMCs cocultured with EmCaMSCs in different ratios (EmCaMSCs:PBMC). PBMCs cultured alone (0:1, 0:2, 0:4, 0:8) were used as a control. (C) The proliferation of PBMCs cocultured with EmCaMSCs and interferon-γ. (D) The fold change in the proliferation of PBMCs cocultured with EmCaMSCs plus nivolumab and/or anti-PD-L1 antibodies. EmCaMSC2 and EmCaMSC3 were used in the experiments, which were conducted in triplicate. *P < 0.05, ***P < 0.01, ***P < 0.001.
EmCaMSC-Secreted IL-8 and IGFBP6 Could Suppress PBMC Proliferation
To further investigate the other mechanisms, we tested the composition of EmCaMSC-CM and found that IGFBP2, IGFBP4, IGFBP6, IL-8, OPG, TIMP-1, and TIMP-2 were highly expressed from ~2- to 3-fold (Fig. 5A and B). The ELISA results confirmed IL-8 (Fig. 5C), IGFBP2 (Fig. 5D), and IGFBP6 (Fig. 5E) concentrations in the CM of various EmCaMSCs (Table 2). EmCaMSCs could secrete a considerable amount of the above three cytokines when compared with the control medium. Furthermore, we wanted to determine the effects of these cytokines on PBMC proliferation when cocultured with EmCaMSCs. We found that IL-8 could decrease the proliferation of PBMCs, and inhibition of IL-8 would increase the proliferation of PBMCs when cocultured with EmCaMSCs (Fig. 5F). Besides, inhibition of IGFBP6, but not IGFBP2, could also increase the proliferation of PBMCs when cocultured with EmCaMSCs (Fig. 5G). Moreover, we tested the effects of these cytokines and anti-PD-L1 separately or in combination on the proliferation of CD4 and CD8 cells when cocultured with EmCaMSCs. The results showed that the combination of three cytokine antibodies could significantly recover CD4 and CD8 cell percentages (Fig. 5H). However, separately adding a single cytokine antibody did not have a recovery effect on T cells (Fig. 5H). Furthermore, adding anti-PD-L1 alone or in combination with anti-IL-8 did not have a recovery effect on T cells (Fig. 5H).

Identification of cytokines from endometrial cancer–derived mesenchymal stem cell–conditioned medium (EmCaMSC-CM) and inhibition of IL-8 and IGFBP6 rescued the proliferation of peripheral blood cells (PBMCs) cocultured with EmCaMSCs. (A) The proteins secreted to the medium from EmCaMSCs were analyzed using a cytokine array. The red boxes indicated the higher protein expression, including IGFBP2, IGFBP4, IGFBP6, IL-8, OPG, TIMP-1, and TIMP-2, in EmCaMSC-CM when compared with the control. (B) The quantification results of fold change in each protein. The concentration of (C) interleukin-8, (D) IGFBP2, and (E) IGFBP6 secreted from different EmCaMSCs was measured. (F) The fold change in the proliferation of PBMCs cocultured with EmCaMSCs plus IL-8 and its blocking antibodies. (G) The fold change in the proliferation of PBMCs cocultured with EmCaMSCs plus anti-IGFBP2 and IGFBP6. EmCaMSC2 and EmCaMSC3 were used in the experiments, which were conducted in triplicate. (H) The percentage of CD4 and CD8 T-cell population from PBMC alone or cocultured with EmCaMSCs and anti-CD3/CD28 stimulation for 3 days is shown. The antibodies were additionally added to the culture as indicated. *P < 0.05, **P < 0.01, ***P < 0.001.
Concentrations of Various Cytokines of EmCaMSCs.

EmCaMSC: endometrial cancer–derived mesenchymal stem cell; SD: standard deviation.
These results demonstrated that EmCaMSCs could secrete large amounts of the above three cytokines, and they suppress the proliferation of PBMCs through paracrine IL-8 and IGFBP6. The combination of three cytokine antibodies could significantly recover CD4 and CD8 cell percentages when cocultured with EmCaMSCs.
EmCaMSCs Suppressed the Cytotoxicity of Cytokine-Induced Killer Cells
Finally, we evaluated the number of cytokine-induced killer (CIK) cell colonies formed after coculturing with EmCaMSCs and after culturing alone. We treated PBMCs and the cells being cocultured with IL-2 (3,000 U/ml) for 4 days. We found that the number of colonies (Fig. 6A) and the expression of CD4, CD8, and CD56 (Fig. 6B) were lower in the coculture group. Subsequently, the IL-2-treated PBMCs (effector cells) were further cocultured with K562 cells (target cells) at effector cell-to-target cell ratios of 5:1 and 10:1 for 4 h. The proportion of apoptotic K562 cells (PI-positive) was significantly lower in the coculture group (Fig. 6C). PBMC killing ability of RL95-2 and HEC-1A (endometrial cancer cell lines) was significantly attenuated after coculture with EmCaMSCs (P < 0.001, Fig. 6D). These results demonstrated that EmCaMSCs could suppress the cytotoxicity of IL-2-induced killer cells by decreasing the number of killer cells and their killing capability.

The cytotoxicity of interleukin-2-induced peripheral blood cells (PBMCs) when cocultured with endometrial cancer–derived mesenchymal stem cells (EmCaMSCs). (A) The morphology of PBMCs cocultured with EmCaMSCs and interleukin-2 (3,000 U/ml; Gibco) for 4 days. PBMCs cultured alone served as a control. (B) The protein expressions of CD56, CD4, and CD8 were analyzed by fluorescence-activated cell sorting. (C) The cytotoxicity assay, analyzing PBMCs killing K562 cells, was based on positive staining by propidium iodide. The effector (PBMC)/target cell (K562) ratio is shown. EmCaMSC2 and EmCaMSC3 were used in the experiments, which were conducted in triplicate. (D) The lactate dehydrogenase (LDH) cytotoxicity assay analyzing PBMCs (cocultured with or without EmCaMSCs) killing RL95-2 and HEC-1A endometrial cancer cells at a 10:1 ratio. **P < 0.01, ***P < 0.001.
Discussion
In this study, for the first time, we successfully isolated CaMSCs from an endometrial tumor. Like BMSCs, these EmCaMSCs also displayed the ability to differentiate into adipocytes, osteoblasts, and chondrocytes. EmCaMSCs were able to inhibit the proliferation of PBMCs and CIK cell cytotoxicity; this indicated that EmCaMSCs may have immunosuppressive effects, which contribute to tumor progression and the failure of immunotherapy. Therefore, an understanding of the characteristics and functions of EmCaMSCs may promote the development of novel therapeutic strategies for the treatment of endometrial cancers.
The cancer microenvironment contains numerous cell types, including endothelial cells, lymphatic circulation, fibroblasts, and various cells derived from bone marrow, such as MSCs and macrophages 15 . Tumor cells can secret chemokines and growth factors to attract MSCs to the tumor sites. Tumor MSCs can secrete cytokines and chemokines, which can affect tumor growth and metastasis16,17. Therefore, crosstalk between various cells (MSCs, tumor cells, surrounding host tissue) in the tumor microenvironment will contribute to tumor progression and evolution 18 . Tumor MSCs can be a therapeutic target. In our study, CaMSCs were found to have an immunosuppression effect in the tumor microenvironment. If immunomodulation medications are used, then tumors can be killed by the patient’s own immune cells. Despite the therapeutic value of tumor MSCs, the percentage of tumor MSCs might predict the prognosis of the patients.
Previous studies reported that tumor MSCs secreted CCL5, which can stimulate breast cancer cell migration, invasion, and metastasis 16 and stimulate PD-L1 expression to suppress immune response 19 . When tumor cells are mixed with MSCs (adult or fetal origin), tumor growth is promoted in vivo more than without mixing with MSCs 17 . BMSCs can promote cell proliferation of multiple myeloma through PD-1/PD-L1 inhibition of immune response 20 . Sun et al. 11 showed that gastric cancer MSC-derived IL-8 could induce PD-L1 expression in gastric cancer cells via the STAT3/mTOR-c-myc signaling pathway. These authors found that block IL-8 derived from MSCs may overcome immune escape induced by PD-L1. Enhanced PD-L1 expression of cancer stromal cells can inhibit CD8+ T-cell immune response and promote colon cancer 21 . In agreement with previous studies, our study also showed that CaMSCs could inhibit immune response via cytokines (IL-8, IGFBP2, and IGFBP6) and PD-L1, which attenuate the killing abilities of PBMC and T cells.
EmCaMSCs expressed PD-L1 and PD-L2, similar to CaMSC derived from prostate cancer 22 ; the expression of these ligands was further upregulated by treatment with TNFα and IFNγ. The PD-L1 promoter has an interferon regulatory factor 1 response element comprising an IFN-γ binding site 23 . BMSCs could inhibit lymphocyte proliferation through the PD-1 pathway 24 . Similar to BMSCs, adipose tissue–derived MSCs can also inhibit T cells via nuclear factor-κb (NFκb) activation, mediated by the PD-L1/PD-1 and galectin-9/TIM-3 pathways 25 . In the present study, we showed that IFNγ could enhance the EmCaMSC-mediated inhibition of PBMC proliferation. However, we found that the inhibition of PD-L1 and PD-1 by anti-PD-L1 antibodies and nivolumab only partially reversed PBMC proliferation, suggesting that other factors may also be involved.
To identify additional possible molecules that may be involved in regulating PBMC proliferation, we analyzed the secreted proteins from EmCaMSC-CM and found that the expression of IGFBP2, IGFBP4, IGFBP6, IL-8, OPG, TIMP-1, and TIMP-2 was significantly higher. Other than PD-L1 and PD-1, we determined that PBMC proliferation could be rescued by inhibiting IL-8 and IGFBP6 under the EmCaMSC-PBMC coculture system. Gastric cancer MSCs enhanced PD-L1 expression of gastric cancer cells by secreting IL-8 11 . Although it was reported that IGFBP2 increased PD-L1 expression in melanoma cells 26 and promoted immunosuppression in glioblastoma 27 , we did not find that IGFBP2 could rescue the proliferation of PBMC. We determined that IGFBP6 contributed to the immunosuppressive effects of EmCaMSCs potentially through the IGF-independent pathway 28 . Thus, inhibition of IL-8 and IGFBP6 may attenuate the immunosuppressive effects of EmCaMSCs, and they could be additional targets for the immunotherapeutic regimen with anti-PD-1/PD-L1.
Furthermore, EmCaMSCs suppressed the cytotoxicity of IL-2-induced killer cells, possibly by directly inhibiting the CD56 and CD8 populations, which possess cytotoxic potential. In addition, tumor stromal cells impaired the cytotoxicity of natural killer (NK) cells by reducing the expression of the NK cell activation receptors NKp44 and NKp46 29 . Further studies are needed to elucidate the mechanisms through which EmCaMSCs suppress IL-2-induced killer cell cytotoxicity.
Most studies about CaMSCs have focused on tumor cells and the production of some common cytokines, such as IL-6, showing the altered signaling pathways in different cancers and suggesting that they may have tissue-specific characteristics. Lung squamous cell carcinoma–derived MSCs were shown to block NK cell activity through IL-6 and prostaglandin E2 (PGE2) 30 . A previous study showed that prostate cancer–derived CaMSCs could inhibit T-cell proliferation through the PD-L1/PD-1 pathway 22 . Collectively, it is suggested that CaMSCs regulate immunosuppression through various cytokines and other molecules, altering the efficacy of immunotherapy. However, due to the low proportion (1%–5%) of CaMSCs and their high similarity with normal MSCs 22 , it is important for potential therapies to selectively target CaMSCs or CaMSC-dependent signaling pathways. Previous animal studies involved coinjecting animals with MSCs and cancer cells (1:1 ratio) 31 ; however, this may not represent real-life scenarios, so the results should be re-evaluated.
In conclusion, we successfully isolated EmCaMSCs from patients with endometrial cancer and showed that they exhibited immunosuppressive effects. EmCaMSCs were found to suppress the proliferation of PBMCs via PD-L1, IL-8, IGFBP6, and IL-2-induced killer cell cytotoxicity. Anti-MSC-secreted cytokine therapy, an immune checkpoint inhibitor, or a combination may have therapeutic value against endometrial cancer. Furthermore, the percentage of EmCaMSCs may predict the prognosis of endometrial cancer.
Supplemental Material
sj-docx-1-cll-10.1177_09636897221104452 – Supplemental material for Endometrial Cancer-Infiltrating Mesenchymal Stem Cells Exhibit Immunosuppressive Effects
Supplemental material, sj-docx-1-cll-10.1177_09636897221104452 for Endometrial Cancer-Infiltrating Mesenchymal Stem Cells Exhibit Immunosuppressive Effects by Kai-Hung Wang, Yu-Hsun Chang, Tomor Harnod and Dah-Ching Ding in Cell Transplantation
Footnotes
Author Contributions
KH Wang: conception, study design, data collection, manuscript writing (draft); YH Chang: conception and data analysis; T Harnod: conception and manuscript writing (draft); DC Ding: supervision, manuscript writing (draft), manuscript revision.
Ethical Approval
This study was approved by the Research Ethics Committee of the Hualien Tzu Chi Hospital (IRB 109-257-A).
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Data Availability
All relevant data are reported in the article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by the Buddhist Tzu Chi Medical Foundation (TCMF-EP-108-02 and TCRD110-62).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.