
Abstract
Energy metabolism programming is a hallmark of cancer, and serves as a potent target of cancer therapy. Valproic acid (VPA), a broad Class I histone deacetylases (HDACs) inhibitor, has been used as a therapeutic agent for cancer. However, the detail mechanism about the potential role of VPA on the Warburg effect in breast cancer remains unclear. In this study, we highlight that VPA significantly attenuates the Warburg effect by decreasing the expression of pyruvate kinase M2 isoform (PKM2), leading to inhibited cell proliferation and reduced colony formation in breast cancer MCF-7 and MDA-MB-231 cells. Mechanistically, Warburg effect suppression triggered by VPA was mediated by inactivation of ERK1/2 phosphorylation through reduced HDAC1 expression, resulting in suppressing breast cancer growth. In summary, we uncover a novel mechanism of VPA in regulating the Warburg effect which is essential for developing the effective approach in breast cancer therapy.
Keywords
Introduction
In 2020, breast cancer is the most commonly occurring cancer all over the world 1 . Breast cancer is one of the most important lethal malignancies threatening women’s health worldwide, which is also the leading cause of cancer mortality 1 . Despite remarkable advances have obtained in researches for treating breast cancer, it remains a severe health problem with the abysmal prognosis, due to breast cancer is prone to metastases 2 . Thus, exploring promising strategies to uncover the underlying mechanism of breast cancer development and identifying new targets for breast cancer diagnosis and treatment are essential.
Dr. Otto Warburg demonstrated that cancer cells take more glucose, leading to high rate of robust glycolysis and lactate production even under sufficient oxygen conditions. This phenomenon is widely termed as the Warburg effect, which is a hallmark of cancer 3 . There are growing studies showing that kinds of metabolism enzymes are up-regulated or activated during tumorigenesis and targeting altered energy metabolism is an effective way to treat cancer or reverse drug resistance 4 –10 . Therefore, blocking the over-expression or activity of metabolism enzymes is an intellectual pursuit to develop anti-cancer drugs.
Valproic acid (VPA), a well-established drug in the long-term therapy of epilepsy, was first synthesized by Burton in 1882. Dr. Göttlicher discovered VPA as a Class I selective HDAC inhibitor 11 . Owing to its HDAC inhibiting capability, VPA is reported to play a critical role in anti-tumor activity in multiply cancer types 12 . Accumulating evidences support that VPA contributes to regulating a broad range of biological functions, including suppressing apoptosis, inhibiting cell proliferation, inducing autophagy and attenuating the epithelial-mesenchymal transition (EMT) in cancer cells 13 –17 . However, the anti-tumor effects of VPA on the modulation of the Warburg effect in breast cancer remain under-investigated.
In this study, we discover that VPA suppresses breast cancer growth by inhibiting aerobic glycolysis through effects on PKM2 expression. Specifically, VPA regulates the Warburg effect via attenuating the level of ERK1/2 phosphorylation by targeting HDAC1. Taken together, targeting PKM2 with the HDAC1 inhibitor VPA in the Warburg effect may provide useful insights for the development of promising therapeutic strategies in breast cancer.
Materials and Methods
Cell Culture
MCF-7, MDA-MB-231 and HEK293 T cells were cultured in Dulbecco Modified Eagle Medium (high glucose) (DMEM, Gibco, ThermoFisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS, HyClone, ThermoFisher Scientific), 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2.
Cell Proliferation Assay
MCF-7, MDA-MB-231 or HEK293 T cells were plated in 6-well plates at a density of 5 × 10 4 cells per well for 24 h and treated with the absence or presence of VPA (Sigma-Aldrich, St. Louis, MO, USA) for 1, 2, 3 and 4 days. The cell numbers were counted at indicated days.
Colony Formation Assay
MCF-7 (600 cells/well) or MDA-MB-231 (6000 cells/well) cells were seeded in 6-well plates and cultured in the absence or presence of 2 mM or 4 mM VPA for 14 days. Cells were washed, fixed with methanol and stained with 1% methylene blue. The colony numbers were counted by using ImageJ software.
Western Blot
Western blot analysis was performed according to a standard procedure. Cells were lysed with lysis buffer [1.5 M NaCl, 1 M HEPES (pH 7.0), 0.1 M NaF, 0.1 M Na4P2O7, 0.1 M Na3VO4, 1% NP40, protease inhibitor] on ice for 30 min and then centrifuged at 12000 r/min for 20 min at 4°C. Protein concentration was determined by a BCA protein assay kit (ThermoFisher Scientific). Equal amounts of protein were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto PVDF membranes (Merck Millipore, Darmstadt, Germany). Membranes were blocked with 5% non-fat milk in Tris-buffered saline-Tween (TBST) at room temperature for 1 h and then incubated overnight at 4°C with primary antibodies against HK2 (Cell Signaling Technology, CST, Danvers, MA, USA, #2867), GPI (Proteintech, Rosemont, IL, USA, #15171-1-AP), ALDOA (Proteintech, #11217-1-AP), GADPH (Proteintech, #60004-1-Ig), PGK1 (Abcam, Cambridge, MA, USA, #ab38007), PGAM1 (Proteintech, #16126-1-AP), ENO1 (Proteintech, #11204-1-AP), PKM2 (CST, #4053), LDHA (CST, #3582), ERK1/2 (CST, #4695), p-ERK1/2 (CST, #4370), β-actin (Proteintech, #60008-1-Ig). Membranes were washed with TBST and incubated with corresponding secondary antibodies HRP-linked anti-rabbit IgG (CST, #7074) or HRP-linked anti-mouse IgG (CST, #7076) for 1 h at room temperature. Immunoreactive bands were visualized using an enhanced chemiluminescence reaction kit (Merck Millipore) and were exposed on X-ray films (Kodak, Rochester, NY, USA). Quantification of Western blot by densitometry was assessed by using ImageJ software.
Cell Metabolism Determination
Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were detected using the Seahorse XFe 96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions. Briefly, MCF-7 or MDA-MB-231 cells were treated in the absence or presence of 2 mM or 4 mM VPA for 12 h and then 1 × 10 4 cells were plated into XFe 96-well plates and pre-treated with or without VPA for 12 h. Cells were washed with Seahorse analysis media and incubated in a CO2-free incubator at 37°C for 1 h. ECAR was measured using Glycolysis Stress Test Kit (Agilent). Glucose (10 mM), Oligomycin (1 μM) and 2-DG (100 mM) were injected into each well sequentially at the indicated time points. OCR was measured using Cell Mito Stress Test Kit (Agilent). Oligomycin (1 μM), FCCP (1 μM) and rotenone/antimycin A (0.5 μM) were injected into each well sequentially at the indicated time points. ECAR or OCR values were normalized to the protein concentration. Data were analyzed by Seahorse XFe 96 Wave software.
Lactate Production Assay
MCF-7 cells were seeded in six-well plates and incubated at 37°C overnight. Fresh phenol red-free DMEM (Gibco) without FBS was added to the plate of 50%-confluent cells for 1 h. Aliquots of media from each well were assessed for the amount of lactate present. Cellular lactate production was determined by using the colorimetric-based lactate assay kit (Sigma-Aldrich) following the manufacturer’s instructions and normalized to cell numbers.
Lentivirus Infection and Generation of Stable Cell Lines
shRNA plasmids targeting HDAC1, HDAC2 and HDAC3 were purchased from Transheep Biological Corporation (Shanghai, China). psPAX2, pMD2.G and pLKO-shRNA-expressing plasmids were co-transfected into HEK293 T cells using Polyethylenimine (PolyScience, Niles, Illinois, USA) following the manufacturer’s protocol. 48 h after transfection, supernatant containing the lentivirus was harvested and mixed with 8 μg/mL polybrene then infected into MCF-7 cells. Cells were selected with 2 μg/mL puromycin and collected for Western blot analysis.
Real-Time Quantitative PCR
Total cellular RNA was isolated using TRIzol reagent according the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). RNA quality was assessed on NanoDrop ONE Microvolume UV-Vis Spectrophotometer (ThermoFisher Scientific) and checked using RNase free agarose gel electrophoresis. Total RNA was used as a template to reverse transcribe into cDNA using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Japan), according to the manufacturer’s protocol. Real-time quantitative PCR was performed in triplicates in a 20 µL reaction mixture containing 2× SYBR Green qPCR Master Mix (Bimake, Houston, TX, USA), 0.25 μM of each of the primers and 50 ng cDNA, using a CFX96 Connect Real-Time System instrument (BioRad, Hercules, CA, USA). Thermal cycling was initiated at 95°C for 10 min, followed by 40 cycles consisting of denaturation at 95°C for 15 s and combined annealing step at 60°C for 30 s, extension at 72°C for 30 s. A melting curve analysis was performed by gradually heating the samples from 60°C to 95°C with 0.5°C increment per second while the fluorescence was measured continuously. The relative expression level was calculated with the 2–ΔΔCT method. Data were normalized to reference gene (β-actin) expression and compared to the control group. The primers used for real-time qPCR analysis were as follows: HDAC1 Forward: 5′-GGAAATCTATCGCCCTCACA-3′, Reverse: 5′-AACAGGCCATCGAATACTGG-3′; HDAC2 Forward: 5′-ATGGCGTACAGTCAAGGAGG-3′, Reverse: 5′-TGCGGATTCTATGAGGCTTCA-3′; HDAC3 Forward: 5′-GCAAGGCTTCACCAAGAGTCT-3′, Reverse: 5′-AGATGCGCCTGTGTAACGC-3′; β-actin Forward: 5′-ACGTGGACATCCGCAAAG-3′, Reverse: 5′-GACTCGTCATACTCCTGCTTG-3′.
Xenograft Animal Model
All animal experiments were approved by the Xinhua Hospital Affiliated to Dalian University Animal Care and Use Committee and carried out in accordance with the regulations of the Service of Consumables and Veterinary Affairs-Division of Animal Protection (SCAV-EXPANIM). 10 female BALB/c nude mice were obtained from the Dalian medical university. 2 × 10 6 MDA-MB-231 cells were adjusted in 200 µL PBS and then inoculated into the subcutaneous part of the right breast of mice. Mice were randomized divided into two groups (n = 5). On day 6, The control group administered intraperitoneally injection of DMSO, while the VPA treatment group received intraperitoneal injection of VPA (500 mg/kg) every other day. The tumor growth of mice was monitored by the in vivo imaging system (IVIS) spectrum system (PerkinElmer, Waltham, MA, USA). After 30 days treatment initiation or when the tumor size reached 2 cm in diameter, mice were sacrificed.
Bioinformatics Analysis
Gene expression levels were analyzed by the public Gene Expression Omnibus datasets (GSE9574). The Human Protein Atlas dataset (https://www.proteinatlas.org) was used for immunohistochemical analysis. Correlations between PKM2 and HDAC1 protein expression in breast cancer were assessed by the Gene Expression Profiling Interactive Analysis (GEPIA) database (http://gepia.cancer-pku.cn/detail.php).
Statistical Analysis
All statistical analyses were performed with GraphPad Prism 6.0 (GraphPad Software, LA Jolla, CA, USA). The statistical differences were analyzed by unpaired, two-tailed Student’s t-test or one-way ANOVA analysis. Data from three independent experiments are expressed as the mean ± standard deviation (SD). Results were considered statistically difference when P < .05.
Results
VPA Inhibits Oncogenesis in Breast Cancer Cells in Vitro
VPA was an effective HDAC inhibitor with a potent anti-tumor activity (Fig. 1A). In the present study, we focused on the anti-tumor activity of VPA in breast cancer. As a model system, we chose two different breast cancer subtypes MCF-7 and MDA-MB-231 cell lines. MCF-7 (estrogen-receptor positive) cell line is Luminal A type tumor which has been extensively studied the mechanism of estrogen-stimulated tumor growth. MDA-MB-231 (estrogen-receptor negative, progesterone receptor negative and HER2/neu oncogene negative) cell line which is invasive triple negative breast cancer (TNBC) type tumor and shows much more aggressive and malignant than MCF-7 cells, in consequence, often used in the study of tumor metastasis in breast cancer. We firstly found that VPA treatment remarkably suppressed the proliferation of MCF-7 and MDA-MB-231 cells (Fig. 1B). Additionally, cell proliferation was inhibited in 293 T cells with VPA treatment as well (Supplemental Fig. S1). In the second, VPA treatment attenuated MCF-7 and MDA-MB-231 cells colony formation capability in a dose-dependent manner (Fig. 1C, D). Collectively, these data indicated that VPA inhibited oncogenesis in breast cancer cells in vitro.
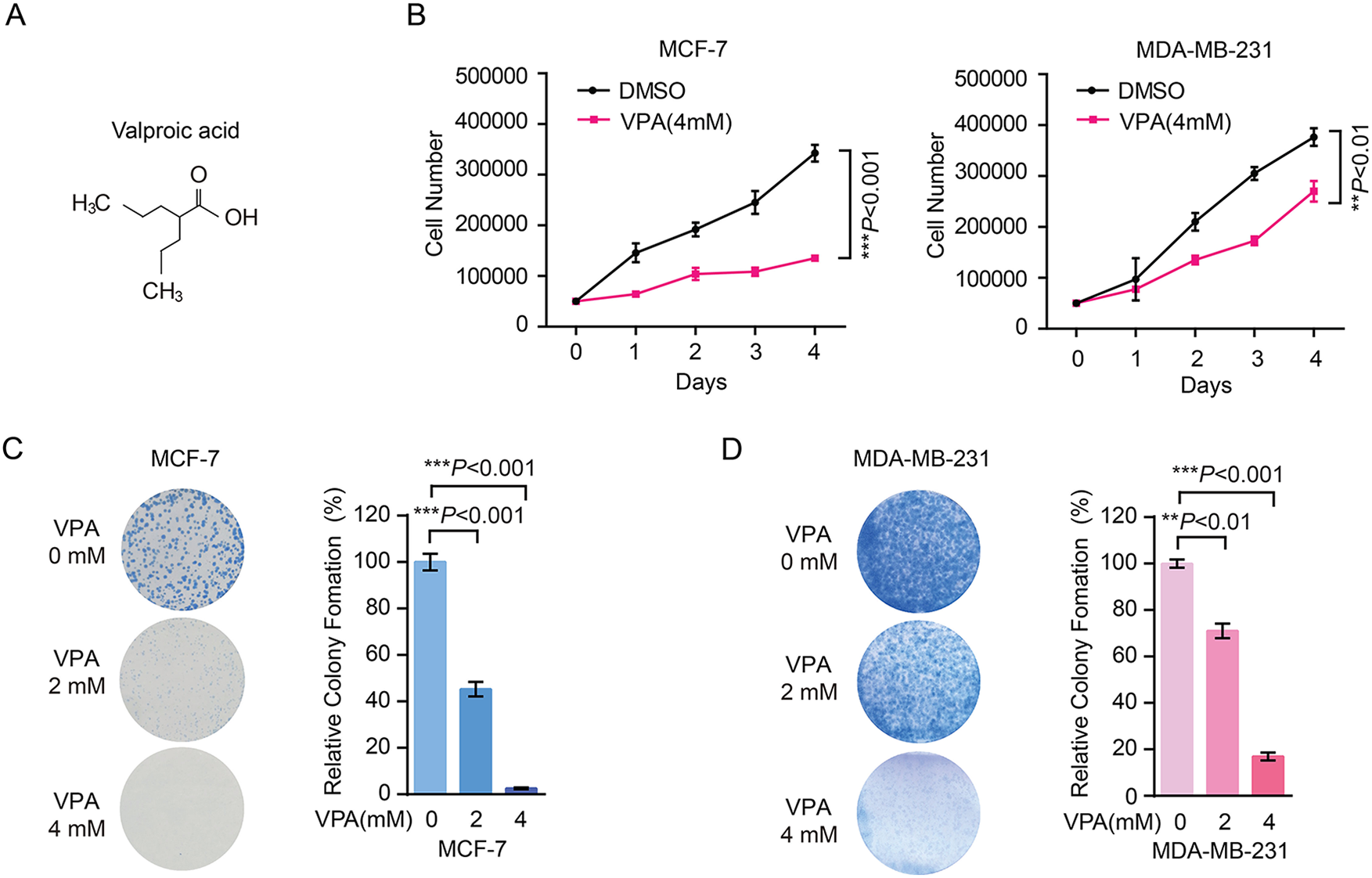
VPA inhibits oncogenesis in breast cancer cells in vitro. (A) Structure of VPA. (B) The cell proliferation of MCF-7 and MDA-MB-231 cells were decreased with 4 mM VPA treatment by cell number counting assay. Student’s t-test was used for statistical analysis. Data represent mean ± SD from three biological replicates (**P < .01; ***P < .001). (C-D) The colony formation of MCF-7 (C) and MDA-MB-231 (D) cells were decreased with 2 mM or 4 mM VPA treatment. Representative images (left panel) and quantification (right panel) of colonies were shown. Statistical significances were calculated by Student’s t-test. Data represent mean ± SD from three biological replicates (**P < .01; ***P < .001).
VPA Inhibits Breast Cancer Growth in Vivo
Based on the in vitro results, we subcutaneously injected MDA-MB-231 cells into nude mice and tumors were imaged after 30 days implantation and dosing. In contrast, the mice in VPA group displayed weaker bioluminescent signal in tumors and the mean tumor size was significantly smaller compared with the DMSO group (Fig. 2A, B). Of note, VPA treatment dramatically retarded tumor growth in vivo. As a consequence, these results strongly illustrated that VPA impaired tumorigenicity of breast cancer cells.
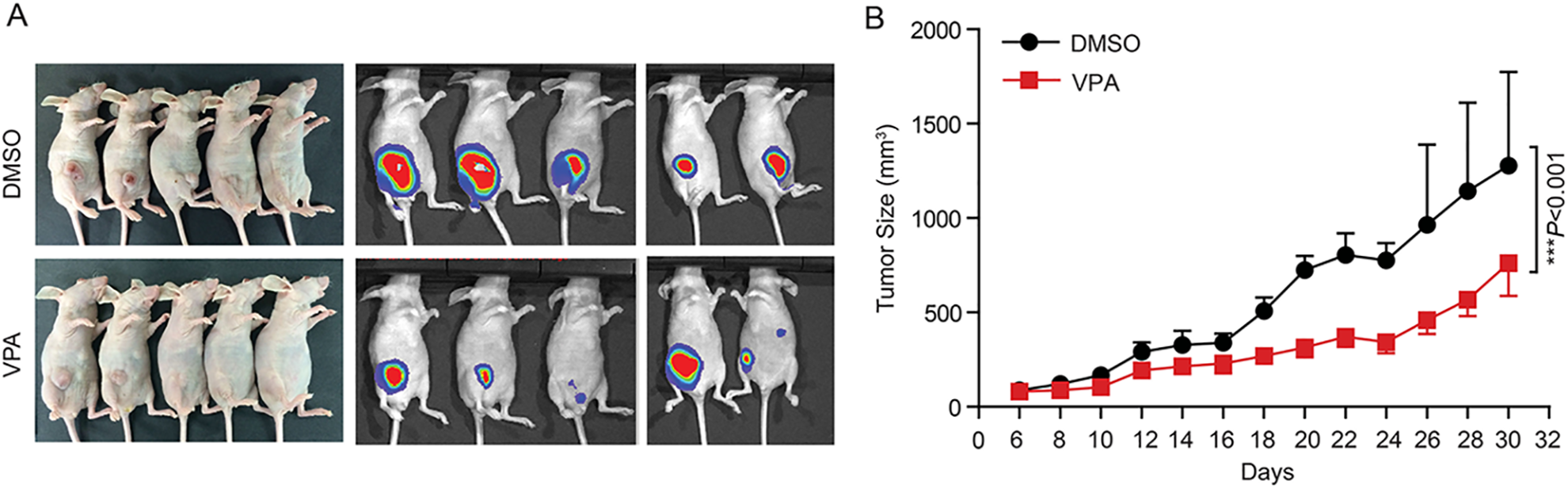
VPA inhibits breast cancer growth in vivo. (A) An IVIS imaging was performed to detect tumor growth in the MDA-MB-231 xenograft nude mice of DMSO group and VPA group. Representative bioluminescent images were shown (n = 5 biologically independent mice per group). (B) VPA reduced primary tumor growth significantly in the MDA-MB-231 xenograft model. Tumor size was measured every 2 days and growth curves were plotted. Statistical significances were calculated by Student’s t-test. Data represent mean ± SD (***P < .001, n = 5 biologically independent mice per group).
VPA Attenuates the Warburg Effect in Breast Cancer Cells
Next, we elucidated the mechanism of VPA inhibiting breast cancer growth from the Warburg effect angle, which played an essential role in regulating cancer cell growth. MCF-7 and MDA-MB-231 cells treated with VPA displayed reduced extracellular acidification rate (ECAR), which was a crucial indicator of aerobic glycolysis (Fig. 3A, B). Furthermore, VPA treatment correlated notably with a decrease of the glycolytic capacity and the glycolytic reserve in MCF-7 and MDA-MB-231 cells, key parameters of glycolytic flux (Fig. 3C, D). Nevertheless, there is still a little difference between MCF-7 and MDA-MB-231 cells about the sensitive of VPA treatment, that may be a predominant result of the different genetic backgrounds between these two cell lines. Subsequently, treatment of MCF-7 cells with VPA led to reduced lactate production (Supplemental Fig. S2A). Additionally, exposure to VPA decreased oxygen consumption rate (OCR) in MCF-7 cells, which stood for mitochondrial respiration (Supplemental Fig. S2B, C). These results suggested that VPA treatment attenuated aerobic glycolysis and oxidative phosphorylation in breast cancer cells.
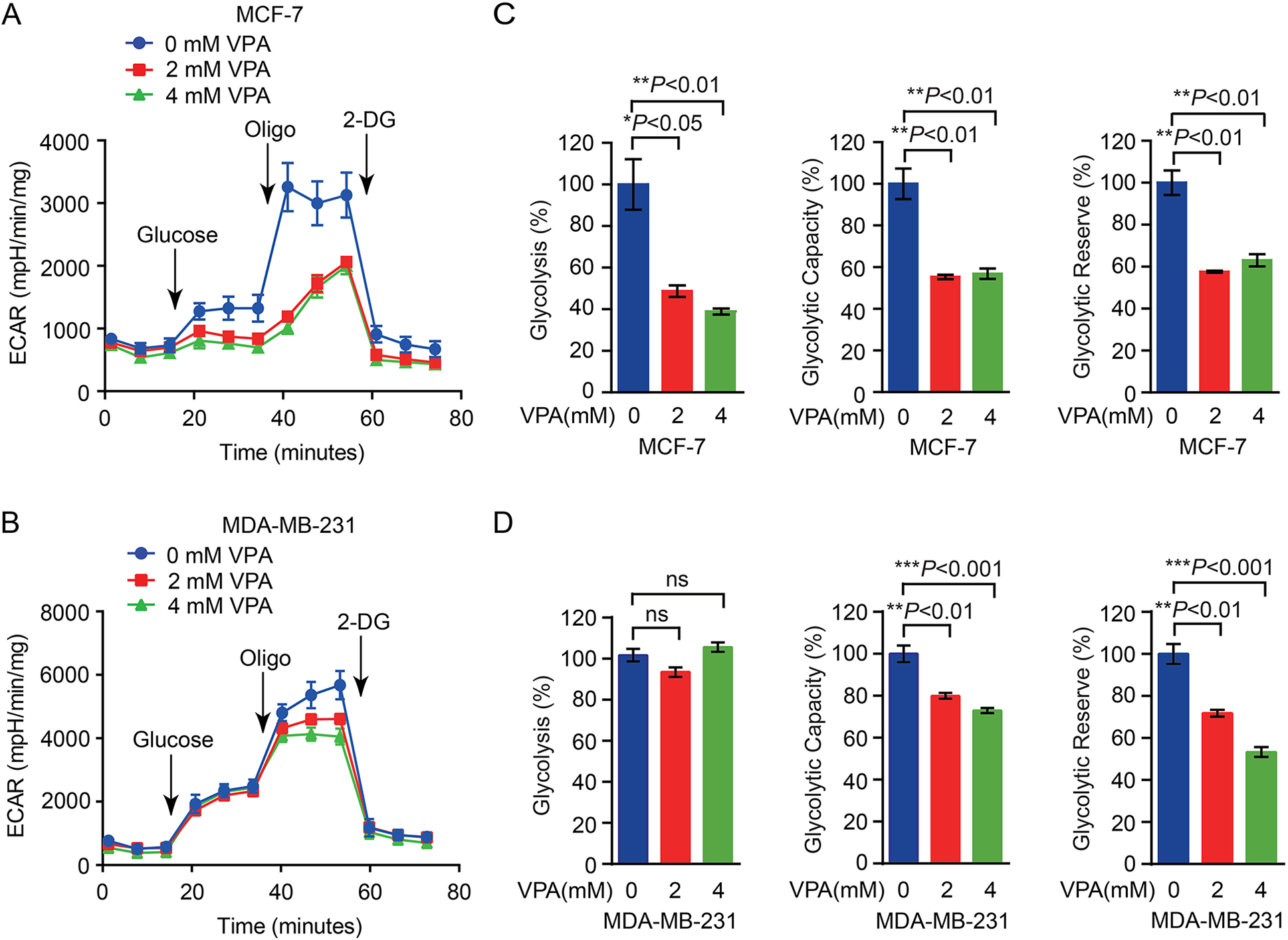
VPA attenuates the Warburg effect in breast cancer cells. (A-B) The glycolysis rate were decreased in MCF-7 (A) and MDA-MB-231 (B) cells with 2 mM or 4 mM VPA by ECAR (a proxy for the rate of glycolysis) analysis. Data represent mean ± SD from three biological replicates. (C-D) The parameters of glycolysis, glycolytic capacity and the glycolytic reserve were tested in MCF-7 (C) and MDA-MB-231 (D) cells with or without VPA treatment. Statistical significances were calculated by Student’s t-test. Data represent mean ± SD from three biological replicates (*P < .05; **P < .01; ***P < .001; ns, not significant).
VPA Affects the Warburg Effect through Down-Regulated PKM2 Expression via Inhibiting ERK1/2 Mediated by HDAC1
To gain further insights into the mechanism underlying VPA in regulating aerobic glycolysis, we determined the expression of a number of key enzymes in the glycolysis pathway. VPA treatment decreased PKM2 protein expression in MCF-7 and MDA-MB-231 cells (Fig. 4A), indicating that VPA repressed glycolysis in breast cancer cells by reducing the expression of PKM2. Moreover, the levels of extracellular signal-regulated kinase (ERK1/2) phosphorylation were significantly decreased when MCF-7 and MDA-MB-231 cells were treated with VPA (Fig. 4B). Importantly, we also observed that PKM2 expression was reduced by ERK1/2 inhibitor U0126 treatment in concentration-dependent manners in MCF-7 and MDA-MB-231 cells (Fig. 4C). Similar results were observed in HEK293 T cells (Supplemental Fig. S3). These results were in line with the research showing that ERK1/2-dependent nuclear PKM2 promoted the Warburg effect in brain tumor 18 . It has been reported that VPA mainly targets to HDAC1, HDAC2, and HDAC3. To better understand the molecular mechanism of VPA in regulating PKM2 expression mediated by ERK1/2 activity, we generated HDAC1, HDAC2 and HDAC3 stable knockdown cells in MCF-7 cells. The knockdown efficiencies of HDAC1, HDAC2 and HDAC3 were confirmed by RT-qPCR (Fig. 4D). We determined the ERK1/2 phosphorylation levels and PKM2 expression in HDAC1, HDAC2 and HDAC3 knockdown MCF-7 cells with VPA treated. Principally, HDAC1 knockdown in MCF-7 cells blocked the effect of VPA on ERK1/2 phosphorylation while decreasing PKM2 expression (Fig. 4E). Collectively, these results explained that VPA regulated PKM2 expression triggering Warburg effect through ERK1/2 signaling pathway targeting HDAC1.
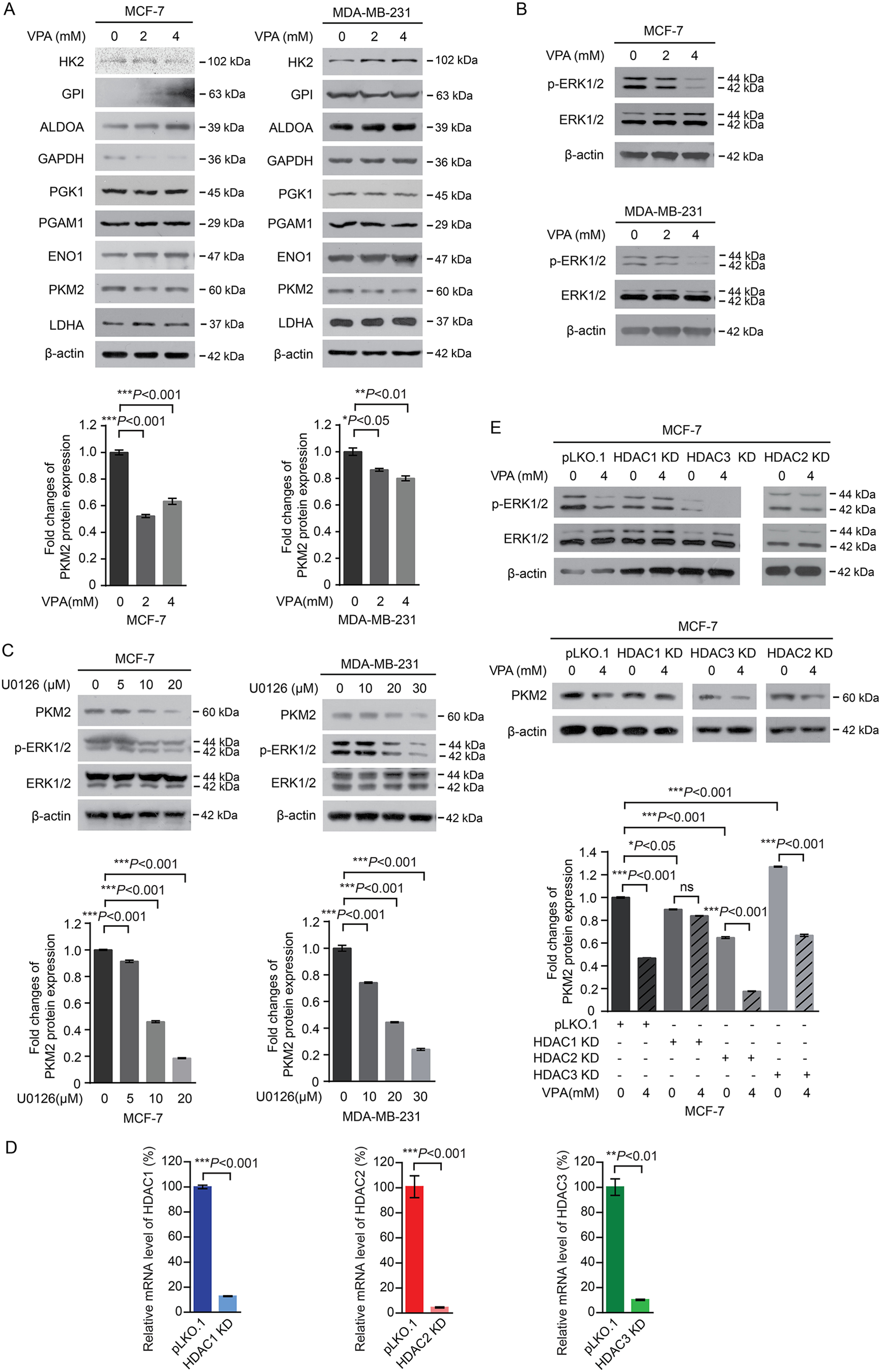
VPA affects the Warburg effect through down-regulated PKM2 expression via inhibiting ERK1/2 mediated by HDAC1. (A) The expression of glycolytic enzymes in MCF-7 and MDA-MB-231 cells untreated or treated with 2 mM or 4 mM VPA were determined by Western blot assay (top panel). Densitometry analysis was performed to assess the PKM2 expression (normalized to β-actin expression) and presented as fold changes over untreated cells (bottom panel). Statistical significances were calculated by Student’s t-test. Data represent mean ± SD from three biological replicates (
PKM2 and HDAC1 Expression are Elevated in Breast Cancer
We then sought to further decipher the role of PKM2 in breast cancer progression. By interrogating the Gene Expression Omnibus (GEO) profiles (GSE9574), we revealed remarkably higher expression of PKM2 in cancer tissues compared with the normal tissues (Fig. 5A). Besides, HDAC1 expression was higher in cancer tissues than in non-tumor tissues but not HDAC2 and HDAC3 (Fig. 5B–D). To gain additional clinical insights, immunohistochemical analyses were assessed by the Human Protein Atlas dataset. Consistently, higher PKM2 and HDAC1 expression levels were also observed in breast cancer tissues (Fig. 5E). In addition, higher PKM2 protein expression was positively correlated with HDAC1 expression in breast cancer from GEPIA database (Fig. 5F). Together, these results demonstrated that targeting HDAC1 by VPA impaired Warburg effect via down-regulating PKM2 expression mediated by ERK1/2 inactivation, thus inhibiting breast cancer cell growth (Fig. 5G).
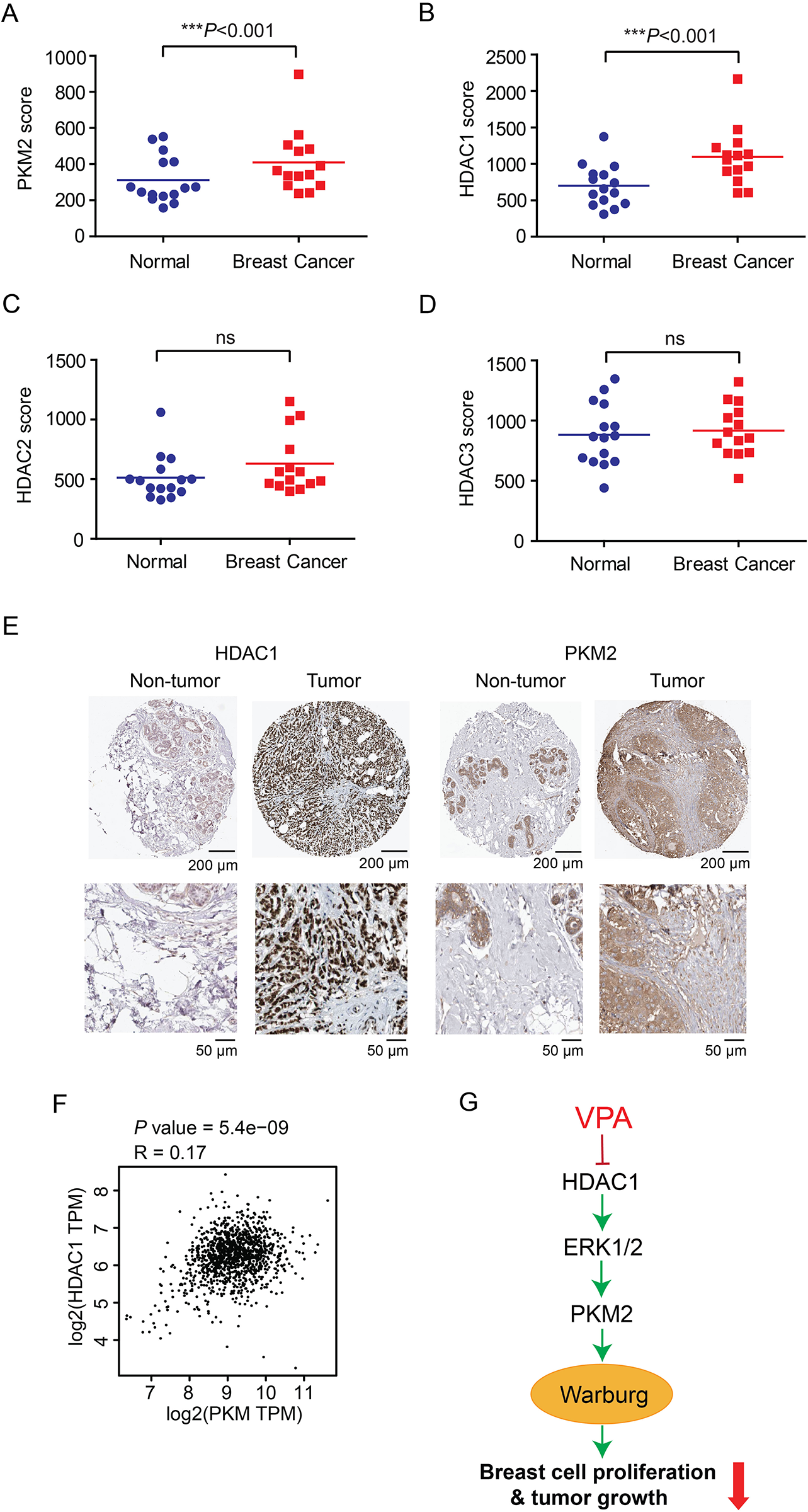
PKM2 and HDAC1 expression are elevated in breast cancer. (A-D) Gene expression levels of PKM2 (A), HDAC1 (B), HDAC2 (C) and HDAC3 (D) in breast cancer tissues and normal tissues, calculated from GEO profiles (GSE9574, normal number = 15, breast cancer number = 14). Student’s t-test was used for statistical analysis (***P < .001; ns, not significant). (E) Representative IHC images showed the increase of PKM2 and HDAC1 expression in breast tumor tissues compared to normal breast tissues from the Human Protein Atlas datasets (scale bar: original magnification 200 μm, inset magnification 50 μm). (F) Scatterplot showing the positive correlation between PKM2 and HDAC1 expression in breast cancer from GEPIA database. (G) The schematic model shows that the role and mechanism of VPA in regulating breast cancer growth.
Discussion
The strategy of “rediscovery of old drugs for new uses,” which possesses merits of reducing the cost and shortening the time of drug development and preclinical research, has been widely concerned over the past decade and applied in systems pharmacology and clinical practice. A case in point is VPA, which is used for epilepsy therapy, while it recently has been validated to have potent efficacies against tumor. Cancer metabolism has gained increasing attention in recent years. The Warburg effect enhances glucose uptake and produces ATP quickly through aerobic glycolysis to meet the energy and biosynthesis demands for rapid cancer cell growth. Emerging studies have focused on targeting metabolism enzymes in cancer treatment, which seems appealing at first glance 9,19 . As a glycolytic enzyme, pyruvate kinase (PK) is a critical rate-limiting mediator of the Warburg effect that catalyzes the irreversible transfer of phosphate from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP) and produces pyruvate and ATP. Among four isoforms of PK, M2 isoform (PKM2) is exclusively expressed in most tumor cells, identifying PKM2 as an exciting avenue for cancer therapy 20,21 . In this study, we demonstrated that PKM2 was generally up-regulated in breast cancer according to the clinical bioinformatics data. VPA treatment could block cell proliferation and tumor growth in breast cancer cell lines. Importantly, VPA suppressed the aerobic glycolysis and tumor progression in breast cancer by decreasing the expression of PKM2. It is particularly noteworthy that the low enzymatic activity of PKM2 is required for the Warburg effect 22 . Further investigation about PKM2 enzymatic activity involved in the regulation of VPA on Warburg effect is needed to refine our understanding. Overall, our study indicates that VPA is an anti-proliferative agent for Warburg effect. Notably, it was recently reported that the inhibition of glycolysis decreased the cancer stem cells population in tumor niche and thereby reduced tumor resistance to therapies 23 . Taken together, targeting Warburg effect by VPA seems to be an effective therapeutic strategy against breast cancer which is highly desirable.
Histone deacetylation is a common epigenetic modification and acts a pivotal part in gene expression and signal transduction, which recently has been exploited as prime anti-cancer drug target. VPA is a HDAC inhibitor, but the signaling pathway by which VPA modulates has not been elucidated, especially in cancer metabolism. ERK1/2 signaling pathway is strongly implicated in cancer. ERK1/2 activation catalyzes the phosphorylation of downstream transcription factors and regulatory molecules rapidly in response to stimulations, which is vital for various cellular activities and physiological processes such as cell cycle progression, cell survival, proliferation, differentiation and metabolism 24 . Here, we discovered that the expression of HDAC1 was elevated in breast tumor tissues and the higher PKM2 expression was positively related to the HDAC1 expression in breast cancer. Moreover, the reduction in HDAC1 expression by VPA may inactivate the ERK1/2 signaling pathway and combine with decreased PKM2 expression, resulting in suppressing the Warburg effect (Fig. 5G). It helps to understand the molecular mechanism by which signaling pathway VPA regulates in the Warburg effect and also gives rise to the role that VPA plays in breast cancer therapy.
Conclusion
Collectively, our data indicated that HDAC1 inhibitor VPA could reprogram the Warburg effect and suppress cancer growth through the negative regulation of PKM2 expression mediated by ERK1/2 signaling pathway.
It is conceivable that VPA might be a therapeutically effective drug targeting glycolysis metabolism against breast cancer, which is expected to have important therapeutic implications.
Supplemental Material
Supplemental Material, sj-jpg-1-cll-10.1177_09636897211027524 - Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect
Supplemental Material, sj-jpg-1-cll-10.1177_09636897211027524 for Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect by Zhen Li, Lina Yang, Shuai Zhang, Jiaqi Song, Huanran Sun, Changliang Shan, Dan Wang and Shuangping Liu in Cell Transplantation
Supplemental Material
Supplemental Material, sj-jpg-2-cll-10.1177_09636897211027524 - Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect
Supplemental Material, sj-jpg-2-cll-10.1177_09636897211027524 for Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect by Zhen Li, Lina Yang, Shuai Zhang, Jiaqi Song, Huanran Sun, Changliang Shan, Dan Wang and Shuangping Liu in Cell Transplantation
Supplemental Material
Supplemental Material, sj-jpg-3-cll-10.1177_09636897211027524 - Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect
Supplemental Material, sj-jpg-3-cll-10.1177_09636897211027524 for Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect by Zhen Li, Lina Yang, Shuai Zhang, Jiaqi Song, Huanran Sun, Changliang Shan, Dan Wang and Shuangping Liu in Cell Transplantation
Footnotes
Abbreviations
Ethical Approval
Ethical approval was obtained for all experimental procedures from the Xinhua Hospital Affiliated to Dalian University Animal Care and Use Committee, Liaoning, China.
Statement of Human and Animal Rights
All procedures in this study were conducted in accordance with the Xinhua Hospital Affiliated to Dalian University Animal Care and Use Committee, Liaoning, China and carried out in accordance with the regulations of the Service of Consumables and Veterinary Affairs-Division of Animal Protection (SCAV-EXPANIM).
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Startup Fund for Distinguished Scholars from Dalian University for Dr. Shuangping Liu, National Nature Science Foundation of China (31560312, 811672781, 81973356 and 81902826), the Program from the Science and Technology Department of Guangdong Province of China (Grant 2017A030313890), the Science and Technology Program of Guangzhou (Grant 201807010003), and the Program of Introducing Talents of Discipline to Universities (111 Project, No. B16021). This work was also supported by grants from the Startup Fund for Distinguished Scholars from Nankai University (63206054 and 91923101) for Dr. Changliang Shan and the National Key R&D Program of China (No. 2018YFC2002000).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.