
Abstract
Islet transplantation is a promising β-cell replacement therapy for type 1 diabetes, which can reduce glucose lability and hypoglycemic episodes compared with standard insulin therapy. Despite the tremendous progress made in this field, challenges remain in terms of long-term successful transplant outcomes. The insulin independence rate remains low after islet transplantation from one donor pancreas. It has been reported that the islet-related inflammatory response is the main cause of early islet damage and graft loss after transplantation. The production of interleukin-1β (IL-1β) has considered to be one of the primary harmful inflammatory events during pancreatic procurement, islet isolation, and islet transplantation. Evidence suggests that the innate immune response is upregulated through the activity of Toll-like receptors and The NACHT Domain-Leucine-Rich Repeat and PYD-containing Protein 3 inflammasome, which are the starting points for a series of signaling events that drive excessive IL-1β production in islet transplantation. In this review, we show recent contributions to the advancement of knowledge of IL-1β in islet transplantation and discuss several strategies targeting IL-1β for improving islet engraftment.
Introduction
Islet transplantation is one of the most promising ways to cure type 1 diabetes by achieving sustained insulin independence and good glycemic control 1,2 . However, clinical islet transplantation faces two major problems before large-scale application: efficacy of restoring insulin production and donor shortages. A successful islet transplantation usually requires multiple pancreatic donors and multiple islet infusions, because islets are particularly vulnerable in the first few days after transplantation 3 . Early stress damage to islet transplantation by various stress stimuli such as inflammation, hypoxia, and immune response has hindered long-term good results of islet transplantation 4,5 . Studies have shown that the loss of islets after transplantation is mainly due to inflammatory events, which occur before adaptive immune-induced rejection, involving the production of cytokines, platelet activation, coagulation and complement system activation, and recruitment of immune cells 6 . Innate immunity is considered to be one of the most important factors affecting early islet loss 7 . The innate immune system is primarily regulated by Toll-like receptors (TLRs) and NOD-like receptors (NLRs), which are pattern recognition receptors that recognize specific danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) and activate nonspecific but powerful inflammatory responses 8 . DAMPs and PAMPs range from bacterial products, adenosine triphosphate (ATP), viruses, and mitochondrial DNA, to granular substances such as crystals and amyloids. This response involves the production of cytokines such as interleukin-1β (IL-1β), which regulate the inflammatory cell recruitment to islets and mediate cytotoxic damage to β cells 9,10 . The amount of pro-inflammatory cytokine IL-1β is upregulated when islets are exposed to warm/cold ischemic stresses during pancreas procurement and to mechanical, enzymatic, and hypoxic conditions during islet isolation and culture (Fig. 1). Moreover, islet resident macrophages, Kupffer cells, and neutrophils around the transplantation site can secrete IL-1β 11 . Islet grafts are exposed to inflammatory damage similar to the early stages of autoimmune diabetes 12 , and IL-1β is one of the key factors in the progression of islet graft failure 13 –17 .

IL-1β increased during pancreas procurement, islet isolation/culture, and islet transplantation. IL-1β: interleukin-1β.
The signaling pathway from TLR activation to mature IL-1β secretion is divided into two steps. First, intracellular signal transduction of TLRs after binding to a ligand is mediated by adaptors such as myeloid differentiation factor 88 (MyD88) 18 . Subsequent activation of the nuclear factor-κB (NF-κB) in the intracellular signaling pathway increases the transcription of the IL-1β gene to encode pro-IL-1β, thereby increasing intracellular pro-inflammatory factor levels. Second, the protein processing of pro-IL-1β is carried out by a complex called inflammasome. Inflammasomes are multiprotein complexes that are activated during infection, inflammation, and autoimmune diseases. These inflammasomes comprise receptors from either the NLR or the ALR family, an adaptor molecule Apoptosis-associated Speck-like protein Containing a caspase recruitment domain (ASC) and the procaspase-1. The NACHT Domain-Leucine-Rich Repeat and PYD-containing Protein 3 (NLRP3) inflammasome has been frequently studied compared to other inflammasomes. The NLRP3 inflammasome comprises a cytoplasmic receptor NLRP3 and ASC that mediates caspase-1-dependent maturation of the pro-inflammatory cytokines IL-1β 19 (Fig. 2). It is worth noting that although there are pathways other than TLRs such as the ATP receptor P2X7 to cause IL-1β production, and other inflammasomes that activate caspase-1, such as NLRP1 and NLRP4, in this review we highlight the roles of the TLRs and NLRP3 inflammasome in islet transplantation. In recent years, researches on TLRs and NLRP3 inflammasome in islet transplantation as well as other organ transplantations have become increasingly important 20 . A systematic understanding of the role of IL-1β, TLRs, and NLRP3 in islet transplantation may improve islet transplantation outcome.
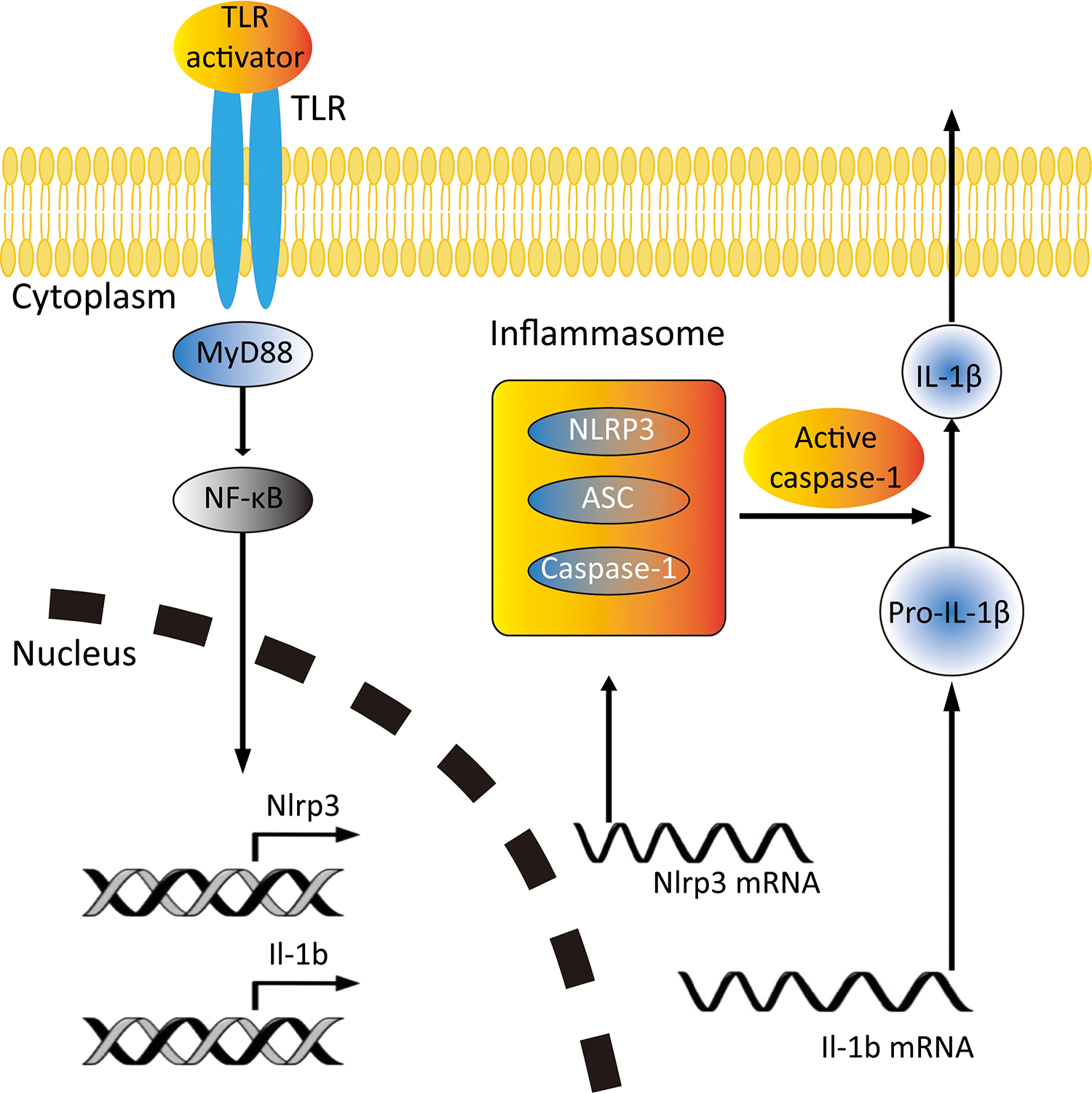
Schematic of TLR and NLRP3 inflammasome activation and IL-1β secretion in islet transplantation. IL-1β: interleukin-1β; NLRP3: NACHT Domain-Leucine-Rich Repeat and PYD-containing Protein 3; TLR: Toll-like receptor.
IL-1β and Islet Transplantation
IL-1β in Pancreas Donors
Although most pancreas and islet grafts are recovered from heart-beating brain-dead (BD) donors, which have much better outcomes than organs from non-heart-beating donors, the islet cells are still damaged from inflammatory events within the donor. Brain death is characterized by extensive cortical necrosis, which can stimulate the production of pro-inflammatory cytokines in a variety of cell types, the so-called “cytokine storm” in BD donors including IL-1β 21 –23 . The release of these pro-inflammatory cytokines has been shown to be associated with brain death, greatly reducing islet yield, functionality, viability, and engraftment post-transplantation 23 . It has been demonstrated that macrophages infiltrate islets during brain death, and that macrophage-related inflammatory cytokines such as IL-1β and IL-6 in islets are induced by brain death 24 .
Recent studies have shown ways to improve islet graft function by inhibiting the BD inflammatory mediator IL-1β before inducing BD. In a BD rat model, administration of exendin-4 to BD donors can increase islet vitality and glucose-stimulated insulin secretion with a significant decrease in IL-1β expression in the pancreas 25 . A selective neutrophil elastase inhibitor, transfusing sivelestat sodium, was shown to decrease IL-1β level and increase islet yield, protect islet function and quality, and inhibit hypercytokinemia-mediated β-cell death in a BD rat model 26 . In a BD nonhuman primate donor model, donor pretreatment with IL-1 receptor antagonist (IL-1Ra) can reduce inflammation by reducing intracellular IL-1β and improve islet survival and engraftment in mice transplanted with minimal islet 27 .
IL-1β in Islet Isolation and Culture
During pancreas digestion and islet purification, islets are exposed to mechanical, enzymatic, ischemic, and osmotic stress. It is well known that enzymes and mechanical stress can induce inflammatory mediators in the islets, such as IL-1β. Gene array study has shown upregulation of genes related to inflammation, apoptosis, cell growth, and angiogenesis immediately after isolation 28 . In that study, IL-1β gene expression was reported to be upregulated by 10 times in islets cultured for 2 days. In cultured adult porcine islets, the presence of mRNA for IL-1β was detected constantly for 11 days and slightly increased during the culture period 29 . It is worth noting that, in the context of clinical islet transplantation, human serum albumin is used instead of fetal bovine serum due to safety issues in using animal-derived materials. Serum deprivation during in vitro culture before transplantation results in islet amyloid polypeptide (IAPP) induced IL-1β secretion and impaired islet vitality and function 30 . Human islets precultured with IL-1Ra showed significant reduction of the pro-inflammatory mediators IL-1β and IL-6 compared to the basal levels, which obviously improved engraftment after transplantation 30,31 .
IL-1β in Inflammatory Response After Islet Transplant
IL-1β is among the key mediators of early islet dysfunction and destruction after islet transplantation, and its role in islet dysfunction has been extensively studied 15 . IL-1β has received special attention due to its important role in the organization and amplification of acute inflammatory responses to tissue damage 32 . In the pancreas, the production and secretion of IL-1β is mainly caused by activated macrophages, neutrophils, endothelial cells, and β cells in islets 33 . Nonspecific inflammation increases IL-1β at the transplant site and affects early graft failure 34 . A syngeneic rat islet transplantation model demonstrated that the gene expression of IL-1β in the islet graft of hyperglycemia rats was significantly increased on the first day after transplantation, compared with the islets before transplantation 35 . On the third day after transplantation, a large number of macrophages existed around the islet tissue and necrotic area, and the apoptosis of β cells was significantly increased compared with the freshly isolated islets. In line with that report, Cardozo et al. found the expression of IL-1β gene in Balb/c islet allograft was detected 8 h after transplantation 36 . In addition, a study suggested a significant increase in IL-1β levels after total pancreatic allograft ischemia-reperfusion injury 37 . Experimental models have shown that inhibition of IL-1β release can reduce islet injury, and circulating anti-IL-1β antibodies can protect islets 38 . Administration of sodium salicylate to inhibit IL-1β-mediated induction of COX-2 can also improve function of pancreatic islets 39 .
IL-1β in Stem Cell–Derived β Cells and Capsules or Devices Encapsulating Islet Cells
Due to islet donor shortages, works on finding alternative resources for β cells are in progress. The development of stem cell–derived β cells such as human embryonic stem cells (hES) and induced pluripotent stem cells (iPSC)-derived insulin-producing cells is expected to offer the solutions. Although several teams have successfully manufactured insulin-secreting cells from hES and iPSC, clinical translation requires new solutions to solve the safety problems regarding teratoma formation and immune rejection 40 . Moreover, stem cell–derived β cells are “immature” that may be more vulnerable to inflammatory response such as IL-1β 41 . These limitations have led to the invention of immune-protected encapsulated devices to contain cells in the device and protect them from attacks of the immune system after transplantation. Many side effects of immunosuppressive agents, including infections, malignancies, worsening renal function, and suppression of islet function, can be overcome by these devices 42 . Studies show capsules or devices encapsulating islet cells can resist to IL-1β damage in the protection of biomaterials 43 . However, the main issues regarding the mass transport of nutrients and oxygen, insulin secretion kinetics, and xenobiotic reactions to encapsulated devices remain to be resolved. Lack of nutrients and oxygen likely results in islet IL-1β production as evidence showed in vitro 30 . In summary, these data indicate that more studies are still required in the field of stem cell–derived β cells and immunoprotective encapsulation strategies.
The Effect of IL-1β on β Cells
Islet β cells are susceptible to IL-1β stimulation during the process of islet transplantation. Through a cascade of intracellular events, low-dose IL-1β leads to a decrease in the biosynthesis and secretion of glucose-stimulated insulin, whereas high-dose IL-1β leads to apoptosis in islets
44
. The binding of IL-1β to the IL-1β receptor (IL-1βR) on the surface of islet cells is the initial step in a series of events leading to gene transcription. After IL-1β interacts with IL-1βR, the receptor undergoes a conformational change, allowing the accessory protein of the IL-1 receptor to be coupled, and subsequently forming a multiprotein complex involving Toll-interacting proteins and the MyD88
45
. This binding results in the activation of tumor necrosis factor receptor-associated factor 6 (TRAF6) by IL-1 receptor-associated kinase (IRAK)
46
. TRAF6 is a type 3 enzyme ubiquitin ligase that mediates the activation of the inhibitor of nuclear factor kappa B kinase (IκK) by ubiquitination, and active IκK phosphorylates the NF-κB inhibitor (IκB), releasing the inhibitory effect on NF-κB and transferring it from the cytoplasm to the nucleus of islet β cell
47
. Traditionally, NF-κB has been broadly referred to as the heterodimer p50/p65 (p50/RelA), an apoptosis-regulating gene
48
. Upon entry into the nucleus, NF-κB induces the expression of inducible nitric oxide synthase in islet β cell. This enzyme acts as a catalyst for
Evidence for Possible Mechanisms of IL-1β Upregulation in Islet Transplantation
Toll-like Receptors
As important innate immune receptors, TLRs are type 1 transmembrane glycoprotein receptors containing leucine-rich repeat motifs that bind various ligands, as well as a highly conserved cytoplasmic domain for the binding of downstream adaptor molecules. TLRs recognize PAMPs, such as bacterial cell membrane lipids, viral double-stranded RNA and DNA oligonucleotides, and can also be activated by host-derived DAMPs such as high mobility groups Box 1 (HMGB1) and heat shock proteins 54 . When islet grafts are isolated and transplanted, many exogenous and endogenous TLR ligands are released 55 . Upon binding to their associated ligand, the TLRs recruit intracellular signaling molecules (e.g., MyD88, Toll interacting protein, IRAK, and TRAF6), resulting in activation of c-Jun N-terminal kinase, p38, and NF-κB signaling pathways 56 . Through these signaling pathways, TLRs activate transcription factors, thereby increasing the expression of various pro-inflammatory cytokines and chemokines 57 . Although TLRs are most commonly associated with infectious diseases, they have recently been associated with noninfectious diseases such as diabetes 58 . Surgical trauma and ischemia-reperfusion injury result in the release of endogenous TLR ligands can also activate TLRs to mediate the inflammatory response 59 . The expression of TLRs in islets has been studied at the message and protein levels in many studies. TLR 2, 3, and 4 mRNA are readily detected in C57BL/6 mice and human islets 60 , and studies have reported TLR2 and TLR4 expression in rat INS-1 and BRIN-BD11 and mouse MIN6 β cell lines 58,61,62 .
The role of TLRs in islet transplantation has been well studied. Mouse syngeneic islet transplantation shows that deletion of donor TLR4, rather than deletion of transplant recipient TLR4, can reduce early post-transplant inflammatory response and increase graft viability. Compared with wild type (WT) islet engrafted in the liver, the TLR4-/- islet grafts have significant lower IL-1β expression 63 . In the mouse allogeneic islet transplantation model, TLRs in both donor islets and recipients are involved in allograft rejection. In that study, the absence of MyD88 in either the donor or the recipient can reduce perigraft infiltration of inflammatory cells and improve transplant outcome 64 . Another group shows that islet transplantation failure can be triggered by TLR2 and TLR4 signaling, and HMGB1 is a possible early mediator. Subsequent downstream signaling leads to inflammation in the islets, followed by T cell–mediated graft destruction 65,66 . In addition, islet isolation can lead to increased expression of TLR4, which can be reduced by prior exposure of donor islets to carbon monoxide, thereby prolonging the survival of allogeneic islets 67 .
In islet xenotransplantation, TLRs activation in porcine islets induces pro-inflammatory and procoagulant responses leading to xenograft rejection 68 . Studies also show that TLR is an important signal for macrophage-mediated pancreatic islet xenograft recognition and rejection. Macrophages exposed to porcine islets have significant higher levels of TLRs gene expression compared with exposure to allogeneic islets 69 . Loss of MyD88 in TLR signaling also leads to impaired macrophage activation after porcine islet xenografts 69 . Although a number of studies have shown the role of TLR signaling in islet transplantation, its significance for islet graft loss remains controversial. Schmidt et al. found that porcine islet xenograft rejection still exists in MyD88-/- mice 70 . Hutton et al. reported that the absence of TLR4, MyD88, and TLR adaptor molecule Ticam-1 did not significantly improve graft survival compared with WT controls 71 . More precise understanding of the TLRs in islet transplantation is warranted.
NLRP3 Inflammasome
In recent years, researchers have found another family involved in innate immunity: the NLRs. The NLR family has three distinct subfamilies: the NOD, NLRP, and IPAF subfamilies, encoded by at least 22 human genes 56 . As mentioned earlier, the NLRP3 inflammasome is a protein complex comprising NLRP3, ASC, and caspase-1. NLRP3 acts as a receptor that senses the exogenous pathogen and endogenous danger signal in the cytoplasm. It consists of 11 leucine repeats at the C-terminus, a NACHT domain in the middle, and a Pyrin domain (PYD) at the N-terminus. Activation of NLRP3 leads to oligomerization and recruitment of ASC and pro-caspase-1, as well as autocleavage and activation of caspase-1 72 . The adaptor protein ASC has a molecular weight of 21.5 kDa and has 195 amino acid residues. It is an important linker protein that consists of the PYD and the Caspase recruitment domain, connecting NLRP3 upstream and caspase-1 downstream. Caspase-1, also known as IL-1β converting enzyme, is an effector protein of the NLRP3 inflammasome and is responsible for cleaving inactive pro-inflammatory cytokines pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18. There are several PAMPs, DAMPs, and environmental stimulus that can activate NLRP3 73 . Endogenous danger signals such as ATP, uric acid crystals, cholesterol crystals, reactive oxygen species, β-amyloid, extracellular matrix components, and lysosomal lysing components, as well as many kinds of exogenous factors such as viruses, bacteria, and fungi can cause assembly and activation of NLRP3 inflammasome.
Studies have shown that elevated blood glucose is associated with activation of NLRP3 inflammasome in islets, resulting in the production of mature IL-1β 74 –76 . However, the relative contribution of endocrine cell types and resident islet macrophages to islet IL-1β secretion was not investigated in these studies. Because TLRs and IL-1β are upregulated in islet transplantation 77 , NLRP3 inflammasome is likely to be activated in this case. Recently, our team found that hypoxia can enhance LPS-induced inflammatory responses by upregulating NLRP3 expression in mouse β cells 78 . Notably, hypoxia is one of the biggest challenges for the islet graft after islet transplantation especially in encapsulating islet cells. We and another group showed in human and porcine islets that NLRP3 inflammasome is expressed and regulated, and hypoxia can increase the gene expression of NRLP3 and pro-IL-1β and induce production of IL-1β and caspase-1 79,80 . Another study shows NLRP3-deficient islets have ex vivo functions and are resistant to hypoxia-induced cell death, compared with wild-type islets 81 . Besides, evidences show that knocking down ASC in β cells can improve IL-1β-induced cell death and reduce cytokine-induced NO production 82 . These studies suggest islet expressing NLRP3 inflammasome may play a role in graft injury after islet transplantation; however, its role in islet transplantation in vivo has yet to be confirmed.
NLRP3 inflammasome expressed in both islet resident and infiltrated macrophages and other immune cells may significantly contribute to graft dysfunction after islet transplantation. Islet β cells can produce IAPP while secreting insulin. IAPP aggregates were detected in clinical pancreatic islet grafts and it can lead to recurrence of hyperglycemia after transplantation 83 –85 . Its potential role in clinical islet transplant failure has been reviewed elsewhere recently 86 . In macrophages, IAPP acts as a potent second signal to activate NLRP3 inflammasome, resulting in the secretion of IL-1β 87 . In diabetic mice transplanted with human IAPP transgenic islets, the number of macrophages increased, and these macrophages were inhibited by exogenous IL-1Ra 13 . In the liver, which is the site of clinical islet transplantation, the Kupffer cells express abundant NLRP3 inflammasome and contribute to acute liver injury 88 . This may destruct the islets after transplantation. These studies indicate that the IL-1β production of immune cells through NLRP3 inflammasome activation in transplant site can damage the β cells.
Therapeutic Opportunity Targeting IL-1β Biology for Islet Graft Dysfunction
Therapeutics targeting the IL-1 system has been studied extensively for islet graft protection. The ligands and receptors of the IL-1 offer various opportunities for intervention such as IL-1Ra, IL-1RTI-IL-RAcP fusion protein (IL-1 trap), and soluble IL-1 receptor type II (sIL-1RII). IL-1Ra is an endogenous IL-1 inhibitor that binds to IL-1R1 and blocks the recruitment of IL-1 R accessory protein (IL-1RAcP), thereby effectively blocking the activation of the IL-1 receptor 89 . The presence of IL-1Ra in human serum from healthy subjects can prevent systemic effects of moderate upregulation of IL-1 release caused by limited injury. Under homeostatic conditions, there is an equilibrium between IL-1β and IL-1Ra 90,91 . Therefore, when the IL-1Ra content is lowered, there is a potential activity of IL-1β leading to downstream effects. The use of this property of IL-1Ra to develop synthetic IL-1Ra and IL-1 trap as anti-inflammatory tools was approved for the treatment of certain types of arthritis in clinic. Moreover, clinical studies have shown that commercially available IL-1Ra (Anakinra) and IL-1 neutralizing antibody (Canakinumab) can reduce insulin requirements and glycated hemoglobin levels in patients with type 2 diabetes 92,93 . Recently, these relatively new compounds have been studied more frequently in islet transplantation 14,15,30,94,95 .
Treatment of rat islet cells with anakinra prevents pro-inflammatory cytokine-induced cell death 96 . After transplantation of pancreatic islets from hIAPP-expressing transgenic mice into diabetic NOD/SCID mice, implantation of an anakinra-containing micro-osmotic pump in recipients attenuates islet amyloid-induced pro-inflammatory cytokine release and islet graft dysfunction 13 . Donor pretreatment with anakinra reduces inflammation and improves viability, mitochondrial membrane polarity, and islet engraftment in mice transplanted with minimal islet mass 27 . In addition, anakinra can synergize with other anti-inflammatory drugs to prevent islet graft damage. Anakinra enhances the protective effect of the tumor necrosis factor alpha inhibitor etanercept on human islets transplanted in mice 97 . The combination of anakinra and etanercept significantly improved islet engraftment compared to monotherapy-treated mice or controls 98 . In a clinical trial of total pancreatectomy with islet autotransplantation, patients received anakinra and etanercept treatment from the day of transplantation to day 7, showed improved islet engraftment and better transplant outcomes 15 . Pretreatment with anakinra and tocilizumab (a monoclonal IL-6 receptor antibody) significantly improved human islet function compared with controls 31 . Other strategies that increase IL-1Ra can also protect post-transplant islets. In mouse allogeneic islet transplantation, α1-antitrypsin exerts allograft-protective activity through the induction of IL-1Ra both in islets and macrophages 99 . IL-1Ra expressing mesenchymal stem cells (MSCs) can mitigate the cytotoxicity of IL-1β in the islets and improve the transplant outcome 100,101 . IL-1 trap is an alternative choice to block IL-1 signaling. The use of IL-1 trap counteracts the destruction of islet cells and prolongs the survival of transplanted islets in diabetic NOD mice 95 . Besides, soluble IL-1 inhibitor sIL-1RII significantly improved the rates of graft survival in anti-CD4 monoclonal antibody treated diabetic NOD mice 102 . These strategies that directly block the IL-1 system showed potential in improving islet transplant outcome. However, more randomized controlled trials in humans need to be conducted in the future, especially IL-1 inhibiting strategies in the isolation bath of the pancreas and in the islet extraction process.
In addition to IL-1-targeted strategies, TLRs and NLRP3 also present important therapeutic opportunities in the prevention of islet graft dysfunction. Blocking TLR4 using a species-specific anti-TLR4 monoclonal antibody significantly increased the survival rate in both syngeneic and allogeneic islet transplantation 103 . Early blockade of TLR4 by TLR4 antagonists protects islets from sustained sterile inflammation-mediated stress during islet isolation and promotes good transplant outcomes 104 . A cell permeable peptide PTD-dnTLR4 blocks TLR4 signaling in both macrophages and β cells, and prolongs the survival of allografts by inhibiting inflammation and macrophage infiltration 77 . TAK-242, a small molecular inhibitor of TLR4, was recently shown to protect islet grafts. Localized drug delivery of TAK-242 within the islets in transplantation site can promote satisfied transplant outcomes 105 . ODN 1585, a CpG oligonucleotide TLR9 agonist, can enhance the expansion of IL-22 producing CD3-NK1.1+ cells in the liver, resulting in IL-22-mediated prolonged allograft survival and increased islets function 106 . Therapeutics targeting NLRP3 in islet transplantation are rarely reported so far. Glyburide, a therapy currently in clinic to treat T2D, can inhibit the NLRP3 inflammasome resulting in decreased IL-1β levels in human islet treated with LPS+ ATP 79 . Our group have reported that human umbilical cord-derived MSCs can protect porcine islets from hypoxia-induced cell death. This effect is partially mediated by decreasing expression of NLRP3 80,107 . Together, strategies targeting TLRs-NLRP3-IL-1β pathway showed promising improvement in experimental islet transplantation models. But further clinical trial is needed to confirm its effectiveness.
Conclusions
Early islet graft dysfunction and loss of islet mass occurs within a few days after transplantation. IL-1β is one of the most important cytokine mediators of inflammation and plays an important role in the pathogenesis of islet graft dysfunction and may represent an early inflammatory marker of graft failure. Since TLRs and NLRP3 inflammasome has been implicated in the mechanism of islet graft loss, the effect of TLRs and NLRP3 on graft dysfunction is likely to be mediated by IL-1β. Nevertheless, there are still many unsolved questions about the role of TLRs and NLRP3 inflammasome in the destruction of pancreatic islet cells during islet transplantation. Meanwhile, IL-1 inhibitor used in clinical studies to date is not enough to fully reduce the insulin needs indicates other signal pathways may also participate the islet destruction. A further understanding of the crosstalk between TLRs-NLRP3-IL-1β pathway and other signal pathways, and the therapeutic options that target this process may improve the outcome of islet transplantation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant Nos: 81201171, 81671752, 81471715, and 81771827), Hunan Provincial Science and Technology Department major project (Grant No.: 2018SK1020), and Natural Science Foundation of Hunan Province, China (Grant No.: 2017JJ3423).