
Abstract
Particulate matter 2.5 (PM2.5)-induced pulmonary inflammation is an important issue worldwide. NLRP3 inflammasome activation has been found to be involved in pulmonary inflammation development. However, whether PM2.5 induces pulmonary inflammation by activating the NLRP3 inflammasome has not yet been fully elucidated. This study researched whether PM2.5 induces the NLRP3 inflammasomes activation to trigger pulmonary inflammation.
Mice and MH-S cells were exposed to PM2.5, BOX5, and Rapamycin. Hematoxylin and eosin staining was performed on the lung tissues of mice. M1 macrophage marker CD80 expression in the lung tissues of mice and LC3B expression in MH-S cells was detected by immunofluorescence. IL-1β level in the lavage fluid and MH-S cells were detected by enzyme-linked immunosorbent assay. Protein expression was detected by Western blot. Autophagy assay in MH-S cells was performed by LC3B-GFP punctae experiment.PM2.5 exposure induced the lung injury of mice and increased NLRP3, P62, Wnt5a, LC3BII/I, and CD80 expression and IL-1β release in the lung tissues. PM2.5 treatment increased NLRP3, pro-caspase-1, cleaved caspase-1, Pro-IL-1β, Pro-IL-18, P62, LC3BII/I, and Wnt5a expression, IL-1β release, and LC3B-GFP punctae in MH-S cells. However, BOX5 treatment counteracted this effect of PM2.5 on lung tissues of mice and MH-S cells. Rapamycin reversed the effect of BOX5 on PM2.5-induced lung tissues of mice and MH-S cells.PM2.5 activated the NLRP3 inflammasome and IL-1β release in MH-S cells by facilitating the autophagy via activating Wnt5a. The findings of this study provided a new clue for the treatment of pulmonary inflammation caused by PM2.5.
Introduction
Particulate matter 2.5 (PM2.5) is particles suspended in the atmosphere with a diameter of no more than 2.5 μm, which carries harmful substances such as heavy metals.1,2 PM2.5 has the characteristics of small size, large surface area, long propagation distance, and long stagnation time. 3 It can be deposited in the alveoli after entering the body through the respiratory system.4,5 After penetrating the lung–blood barrier, PM2.5 can enter the blood circulatory system to cause damage to the tissues of multiple organs. 6 Particularly, after being inhaled into the lung, PM2.5 can induce pulmonary inflammatory response through inducing the release of multiple inflammatory factors. 3 Meanwhile, the harmful substances (such as polycyclic aromatic hydrocarbons) carried by PM2.5 can cause the production of free radicals in the lung tissues. Free radicals will break the balance of oxidants and antioxidants to induce the lung injury and pulmonary dysfunction.7-9 Therefore, the identification of the molecular mechanism in PM2.5-induced pulmonary inflammation is the key to treat this disease.
Inflammasome is a main regulator of innate immunity, which participates in promoting the inflammatory response by activating caspase-1 and interleukin (IL)-1β. 10 Nucleotide oligomerization domain (NOD)-like receptor protein 3 (NLRP3) inflammasome is an important part of body’s innate immunity. Pathogens, such as viruses, parasites, and bacteria, endoplasmic reticulum stress, and oxidative stress can trigger the activation of the NLRP3 inflammasome. 11 As an intracellular multi-protein complex, NLRP3 inflammasome includes the expression of NLRP3, pro-caspase-1, and apoptotic speck protein. 12 NLRP3 inflammasome can promote the release of inflammatory factors (such as IL-1β) by cleaving the pro-caspase-1 into caspase-1 via formatting an inflammasome complex. Meanwhile, NLRP3 inflammasome can activate the downstream complex signaling pathways to trigger a series of inflammatory reactions.13,14 As a pro-inflammatory cytokine, IL-1β is considered as an effector of the NLRP3 inflammasome, which participates in the process of the PM2.5-induced pulmonary inflammation. 15 Previous study has been reported that PM2.5 could induce the pulmonary inflammation through activating the NLRP3. 3 In the PM2.5-induced mouse lung injury model, PM2.5 has been found to activate the NLRP3 inflammasome to induce the over-production of IL-1β. 16 However, the intrinsic molecular mechanism of PM2.5 activating NLRP3 has not yet been fully elucidated. Thus, this study was designed to research the intrinsic molecular mechanism of PM2.5 on the NLRP3 activation.
Autophagy is an important metabolic process, which maintains the self-stable state of cells by removing the damaged proteins and organelles. 17 Autophagy is involved in a variety of stress responses, such as inflammatory reaction. 18 However, the over-activated autophagy can cause the cell damage and death, thereby triggering a series of inflammatory reactions. 19 It has been revealed that PM2.5 could intensify the autophagy activation and induce the subsequent death of human bronchial epithelium cells. 20 Intriguingly, the inhibition of the autophagy could protect against lung inflammation by suppressing the activation of the NLRP3 inflammasome. 21 However, the enhanced autophagy could activate the NLRP3 inflammasome to augment the lung inflammation. 22 Therefore, this study speculated that PM2.5 might induce the activation of the NLRP3 inflammasome via aggravating the autophagy.
Recently, it has been found that PM2.5 induced the expression of inflammatory cytokines in human bronchial epithelium cells via activating the Wnt5a. 23 Wnt5a played an important role in the autophagy of macrophages. 24 However, whether PM2.5 activates the autophagy via inducing Wnt5a activation has not yet been elucidated. In our preliminary study, we used PM2.5 to treat B lymphocytes (WEHI-231 cells), T lymphocytes (EL4 cells), dendritic cells (JAWSII cells), and alveolar macrophage cells (MH-S cells) derived from lung tissues of mouse and then detect the expression of NLRP3 in the four cell lines. It was found that MH-S cell expressed the highest NLRP3 mRNA than the other three cell lines (as shown in Supplement Figure S1). Thus, this study used PM2.5 to treat C57BL/6 nude mice and MH-S cells. The aim was to verify the following speculation: PM2.5 might promote macrophages autophagy by activating Wnt5a, and then further promote the activation of the NLRP3 inflammasome to induce the pulmonary inflammation. The findings of this paper might provide a novel clue for the treatment of PM2.5-induced pulmonary inflammation.
Materials and methods
Preparation of PM2.5
The PM2.5 sample was collected from the building roof of the Second Hospital of Hebei Medical University. The PM2.5 sampler (air particulate sampler) was placed on the roof of the building from January 3, 2021 to February 3, 2021. The PM2.5 sampler was about 500 m away from the main traffic road with the height of about 35 m. The flow rate of the air particulate sampler was 100 L/min. The sources of PM2.5 were automobile exhaust, soil dust, industrial emissions, etc. That were emitted into the environment. The glass fiber filter paper was utilized for the collection of PM2.5. The PM2.5 was put into a freeze-drying bottle and placed in a refrigerator at −80°C. After being frozen and evacuated, the PM2.5 particles were dried into dry powder. After being sterilized, the PM2.5 dry powder was stored at −20°C. 3
Morphology and main elements detection of PM2.5 by scanning electron microscope and energy dispersive spectrometer
PM2.5 sample was adhered to the double-sided carbon tape, and then fixed on the sample stage. After being gold-coated by a sputter coater (Desk V, Denton Vaccum), the morphology of PM2.5 sample was observed under a SEM (JSM-840, Jeol, Tokyo, Japan). EDS (JED-2300, Jeol, Tokyo, Japan) was applied for the analysis of the main elements in PM2.5. With a heated tungsten filament to be electron source, the images were obtained from PM2.5 sample. The acceleration voltage was 10 kV.
Animals and treatment by PM2.5, BOX5, and rapamycin
All protocols of animal handling and sampling were approved by the Institutional Animal Care and Use Committee of China Medical University (No. CMU2021111). All efforts were made to minimize the suffering of animals according to recommendations proposed by the European Commission (1997). The study was carried out in accordance with the approved protocol. All methods were conducted in accordance with relevant guidelines.
C57BL/6 nude mice (n = 20, 5 weeks old, average weight of [22 ± 3] g) were commercially provided by Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). All mice were housed in a 12 h day/night cycle room (at about 22°C) with free access to food and water. These mice were randomly divided into four groups: control group (n = 5), PM2.5 group (n = 5), PM2.5 + BOX5 group (n = 5), and PM2.5 + BOX5 + Rapamycin group (n = 5).
Before using, the PM2.5 dry powder was dispersed into the sterilized phosphate buffered saline (PBS) to a concentration of 1 mg/mL. 3 For mice of the control group, 10 μL of PBS was dripped into the nostrils. Mice of the PM2.5 group were nasally dripped with 10 μL of the PM2.5 PBS suspension (1 mg/mL). The nasally dripping was performed for 14 consecutive days twice daily (at 8:00 a.m. and 15:00 p.m.). 25
For PM2.5 + BOX5 group, BOX5 (0.5 μg/mL, 10 μL) was nasally instilled into mice for two weeks with twice a week. 26 PM2.5 PBS suspension (1 mg/mL, 10 μL) was then nasally dripped into mice for 14 consecutive days twice daily (at 8:00 a.m. and 15:00 p.m.). For PM2.5 + BOX5 + Rapamycin group, mice were nasally instilled with BOX5 (0.5 μg/mL, 10 μL) for two weeks with twice a week and administered rapamycin (4 mg/kg, dissolved in 0.25% Tween) for 6 consecutive days per week (for 2 weeks) through intraperitoneal injection. 27 PM2.5 PBS suspension (1 mg/mL, 10 μL) was then nasally dripped into mice for 14 consecutive days twice daily (at 8:00 a.m. and 15:00 p.m.).
On the 15 day, mice of each group were euthanized. The method of euthanizing the mice was as follows: Pentobarbital at a dose of 60 mg/kg (P0225, EKEAR Bio, Shanghai, China) was employed to deeply anesthetize the mice through intraperitoneal injection. Mice were considered to be deeply anesthetized if they had no response to head and limb stimulation. Then the mice were sacrificed via rapid cervical dislocation. The trachea and lungs of mice were separated and the right lung was ligated. The upper end of the trachea was inserted with a puncture needle and then ligated. The left lung was washed three times with PBS. The volume of PBS used per time was 0.3 mL. The lavage fluid was collected and centrifuged for 10 min at 1500 r/min/min and 4°C. After centrifugation, the supernatant of the lavage fluid was obtained and stored at −20°C for the following cytokine testing. The lung tissues of all mice were then collected and stored in a refrigerator at −80°C.
Hematoxylin and eosin staining
The right lung tissues of mice were fixed for 48 h in 4% paraformaldehyde. Then tissues were dehydrated by an ascending gradient of ethanol, followed by being embedded into paraffin. The lung tissues were cut into sections by using a rotary microtome to a thickness of 5 μm. Hematoxylin staining solution and eosin staining solution was sequentially utilized to stain the sections. The procedure of staining was performed in line with the directions. An ascending gradient of ethanol was used to dehydrate the sections. After treated by xylene, the sections were sealed in neutral gum and observed under a light microscope (Olympus, Japan).
Cell culture
Mouse alveolar macrophage cell line (MH-S) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). RPMI-1640 medium containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin was used to culture MH-S cells at 37°C, 5% CO2.
Cell counting kit-8 assay
The effect of PM2.5 on MH-S cell viability was explored by CCK-8 assay. MH-S cells were seeded into 96-well plates and cultured by RPMI-1640 medium containing 10% FBS and different concentration of PM2.5 (12.5, 25, 50, 100 and 200 μg/mL). After being cultured for 48 h at 37°C, 5% CO2, MH-S cells were incubated for 2 h with 10 μL of CCK-8 (Solarbio, Beijing, China) solution. The optical density value of each well was then measured using a porous microplate reader (BioTek, Winooski, VT USA). Five repeated wells were set for MH-S cells of each group. MH-S cells culture by RPMI-1640 medium containing 10% FBS were used as the control. The viability of MH-S cells was determined by the formula of (OD value of experimental group/OD value of control) × 100%.
PM2.5, BOX5 and rapamycin treatment of MH-S cells
By ultrasound, the sterilized PM2.5 dry powder was uniformly dispersed into RPMI-1640 medium (10% FBS) to a final working concentration of 100 μg/mL. 28 MH-S cells (1 × 106 cells) were cultured in RPMI-1640 medium (1 mL) containing 10% FBS and 100 μg/mL PM2.5 for 8 h at 37°C, 5% CO2 (used as PM2.5 group). MH-S cells (1 × 106 cells) cultured in 1 mL of RPMI-1640 medium containing 10% FBS for 8 h were set as the control group.
BOX5 was a specific inhibitor of Wnt5a. RPMI-1640 medium containing 10% FBS and BOX5 (100 μM) was used to pre-treat MH-S cells for 1 h at 37°C, 5% CO2. Then MH-S cells were cultured for 8 h with RPMI-1640 medium containing 10% FBS and 100 μg/mL of PM2.5 in the presence of BOX5. 29 These cells were named the PM2.5 + BOX5 group. MH-S cells were pre-treated by RPMI-1640 medium containing 10% FBS, BOX5 (100 μM) for 1 h, and then treated by RPMI-1640 medium containing 10% FBS and Rapamycin (an autophagy activator, 3 μM) for another 1 h. Followed by this, these MH-S cells were cultured with RPMI-1640 medium containing 10% FBS and PM2.5 (100 μg/mL), BOX5 (100 μM), and Rapamycin (3 μM) for 8 h (used as the PM2.5 + BOX5 + Rapamycin group).
Immunofluorescence
The LC3B expression in MH-S cells was detected by immunofluorescence. MH-S cells of each group were fixed by 4% paraformaldehyde for 15 min, followed by being incubated with 0.1% Triton-X for 10 min. Then 1% bovine serum albumin (BSA) was used to block cells for 1 h. MH-S cells were then treated by rabbit anti-LC3B (AF5225, Beyotime, Shanghai, China) antibody for 12 h at 4°C. Alexa Fluor 594-conjugated goat anti-rabbit secondary antibody (ab150080, Abcam, Shanghai, China) was added to incubate cells for 2 h at room temperature. The nuclei were stained by 4′, 6-diamidino-2-phenylindole (DAPI). LC3B expression (red fluorescence) were observed under a fluorescence microscope (CKX41, Olympus, Tokyo, Japan).
The detection of M1 macrophage marker CD80 expression in mice lung tissues was implemented by immunofluorescence. The right lung tissues of mice were fixed for 48 h by 4% paraformaldehyde, and then dehydrated by ascending gradient ethanol. After being embedded into paraffin, the lung tissues were cut into sections (5 μm). The sections were sequentially treated by 0.1% Triton-X for 10 min, 1% BSA for 1 h, and then rabbit anti-CD80 primary antibody (ab215166, Abcam, Shanghai, China) overnight at 4°C. Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (ab150077, Abcam, Shanghai, China) was added onto the sections for 2 h treatment at room temperature. DAPI was dropped onto the sections for the nuclei staining. The expression of CD80 was observed under a fluorescence microscope (CKX41, Olympus, Tokyo, Japan).
Enzyme-linked immunosorbent assay
MH-S cells of each group were collected and incubated by lysis buffer for 30 min on ice. The cell lysate were centrifuged for 15 min at 12,000 r/min/min and 4°C. The supernatant was collected to detect the level of IL-1β by ELISA. Additionally, the supernatant of mice lavage fluid was collected to detect the level of IL-1β by ELISA. The procedure was carried out by using an ELISA kit (CME0015-048, 4A Biotech, Beijing, China) according to the instructions.
Autophagy assay of MH-S cells by LC3B-GFP punctae experiment
MH-S cells of each group were cultured in the 6-well plates (1 × 105 cells per well). The LC3B-GFP virus (10 μL, BacMam 2.0, Thermo Fisher, Shanghai, China) was then added into each well to treat MH-S cells for 24 h. This process was carried out in line with the manufacturer’s protocol. After that, the LC3-GFP positive autophagasome were observed under a fluorescence microscope (CKX41, Olympus, Tokyo, Japan).
The ingestion of PM2.5 into MH-S cells by transmission electron microscope
TEM was utilized to observe the ingestion of PM2.5 into MH-S cells. Briefly, MH-S cells of the control group, the PM2.5 group, the PM2.5 + BOX5 group, and the PM2.5 + BOX5 + Rapamycin group were cultured in the 6-well plates for 8 h with the relevant medium. Then cells were digested with trypsin and washed 3 times by PBS. Cells were fixed by 2.5% glutaraldehyde for 15 min and washed by PBS. Osmium tetroxide was used to fix cells for 90 min. Cells were then dehydrated by using an ascending gradient alcohol and ascending gradient acetone. Thereafter, cells were sealed in neutral resin and cut into slices with a thickness of 70–90 nm. Uranyl acetate and lead citrate were used to stain the sections according to the instructions. The sections were loaded onto copper grid and observed under a TEM (H-7650, Hitachi, Japan).
Western blot
Lung tissues of mice and MH-S cells were treated with RIPA buffer (Solarbio, Beijing, China) for 30 min on ice to extract the total proteins. Protease Inhibitor Cocktail (Solarbio, Beijing, China) was pre-added into the RIPA buffer. The lysate samples were centrifuged at 14,000g and 4°C for 15 min to collect the supernatant. The total proteins in the supernatant underwent the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for the separation. After being transferred onto the polyvinylidene difluoride (PVDF) membranes, the proteins were blocked by 5% skimmed milk for 1 h at room temperature. Afterwards, primary antibodies were used to probe the proteins for 12 h at 4°C. The primary antibodies were rabbit anti-NLRP3 (1:1000, 15101, Cell Signaling Technology, Boston, USA), rabbit anti-P62 (1:1000, 18420-1-AP, Proteintech, Wuhan, China), rabbit anti-LC3B (1:1000, 2775, Cell Signaling Technology, Boston, USA), rabbit anti-Wnt5a (1:1000, MAB645, R&D, Minneapolis, USA), rabbit anti-pro-Caspase-1 (1:1000, ab179515, Abcam, Cambridge, UK), rabbit anti-cleaved Caspase-1 (1:1000, Cell Signaling Technology, Boston, USA), rabbit anti-pro-IL-1β (1:1000, WL02257, Wanlei Biotechnology, Shenyang, China), rabbit anti-pro-IL-18 (1:1000, FNab06765, Fine Biotechnology, Wuhan, China), and rabbit anti-β-actin (1:1000, ab8227, Abcam, Cambridge, UK). After washing twice using tris-buffered saline with 0.5% Tween-20 (TBST), the proteins were incubated with horseradish peroxidase (HRP)-labeled anti-rabbit secondary antibody (1:10,000, AS014, Abclonal, Wuhan, China) for 2 h at room temperature. Enhanced chemiluminescence (ECL) reagent (Solarbio, Beijing, China) was used to develop the proteins blots according to the instructions. The gray value of protein blots was evaluated by using an ECL Western blot detection kit (Amersham, Little Chalfont, UK). The operation was carried out strictly in line with the instructions. The blots were cut prior to hybridization with antibodies during blotting.
Statistical analysis
All experiments were independently repeated three times. Data was presented in the form of mean ± standard deviation. SPSS 19.0 software (SPSS, Inc. Chicago, IL, USA) was used for the data analysis. Two tailed paired Student’s t-test and one-way variance analysis (followed by Tukey’s post hoc test) were used, respectively, for the difference comparison between two groups and in more than two groups. p < .05 meant a statistically significant difference.
Results
PM2.5 induced the lung injury and activated the NLRP3 inflammasome, autophagy, and IL-1β release in lung tissues
The effect of PM2.5 on the lung injury of mice was researched. H&E staining showed that, relative to the control group, lung tissues of mice in the PM2.5 group were severely damaged, as exhibited by the disordered alveolar structure, the thickened alveolar septal and monocyte-macrophage infiltration (Figure 1(a)). Western blot showed that, in comparison to the control group, significantly higher expression of NLRP3, P62, Wnt5a, and LC3BII/I proteins was found in mice lung tissues of the PM2.5 group (p < .05) (Figure 1(b)). ELISA exhibited markedly higher IL-1β level in mice lavage fluid of the PM2.5 group than the control group (p < .05) (Figure 1(c)). The expression of M1 macrophage marker CD80 in mice lung tissues was detected by immunofluorescence to evaluate the M1 polarization of macrophages. Relative to the control group, mice of the PM2.5 group showed the intensified staining of CD80, indicating the increased M1 macrophages in lung tissues of the PM2.5 treated mice (Figure 1(d)). Therefore, PM2.5 treatment induced the lung injury of mice and activated the NLRP3 inflammasome, Wnt5a, autophagy, and IL-1β release in mice lung tissues. PM2.5 induced the lung injury and activated the NLRP3 inflammasome, autophagy, and IL-1β release in lung tissues. (a) Histological changes of the lung tissues by H&E staining. (b) The expression of NLRP3, P62, LC3BII, LC3BI, and Wnt5a proteins in the lung tissues by Western blot. Three Western blots were replicated for the graph. The blots were cut prior to hybridization with antibodies during blotting. (c) IL-1β level in the lavage fluid of mice by ELISA. (d) Immunofluorescence of M1 macrophage marker CD80 in mice lung tissues. * p < .05.
Inhibition of Wnt5a attenuated the PM2.5-induced lung injury in mice
This study used BOX5 (a specific inhibitor of Wnt5a) and Rapamycin (an autophagy activator) to treat the PM2.5-induced mice. The lung tissue injury of mice was detected by H&E staining. As shown in Figure 2(a), no obviously damage was observed in the lung tissues of the control group mice. However, mice of the PM2.5 group showed the severally lung injury, such as the disorganized alveolar architecture, the thickened alveolar septa, and the markedly infiltrated mononuclear macrophages. Relative to the PM2.5 group, mice of the PM2.5 + BOX5 group exhibited much attenuated lung injury, as manifested by the decreased alveolar structural disorder, the decreased alveolar septal thickening and the decreased monocyte-macrophage infiltration. However, Rapamycin treatment reversed the effect of BOX5 on the lung injury in the PM2.5-induced mice. Additionally, Western blot was utilized to monitor the expression of NLRP3, P62, LC3BI, LC3BII, and Wnt5a proteins in lung tissues (shown in Figure 2(b)). Mice of the PM2.5 group expressed distinctly higher proteins of Wnt5a, NLRP3, P62, and LC3BII/I in lung tissues than the control group (p < .05). Intriguingly, mice of the PM2.5 + BOX5 group expressed lower NLRP3, P62, Wnt5a, and LC3BII/I proteins in lung tissues than the PM2.5 group (p < .05). However, in comparison to the PM2.5 + BOX5 group, much higher NLRP3, P62, and LC3BII/I proteins was observed in the lung tissues of the PM2.5 + BOX5 + Rapamycin group mice (p < .05). The expression of Wnt5a protein was much lower in the lung tissues of the PM2.5 + BOX5 + Rapamycin group mice when relative to the PM2.5 group (p < .05). The expression difference of Wnt5a protein in mice lung tissues was not obvious between the PM2.5 + BOX5 group and the PM2.5 + BOX5 + Rapamycin group. The IL-1β level in mice lavage fluid was explored by ELISA, and the results are shown in Figure 2(c). Mice of the PM2.5 group had higher IL-1β level in lung tissues than the control group (p < .05). Conversely, lower IL-1β level was observed in mice lavage fluid of the PM2.5 + BOX5 group when compared to the PM2.5 group (p < .05). Intriguingly, relative to the PM2.5 + BOX5 group, higher IL-1β level was occurred in mice lavage fluid of the PM2.5 + BOX5 + Rapamycin group (p < .05). Inhibition of Wnt5a attenuated the PM2.5 induced lung injury in mice. (a) Histological changes of the lung tissues by H&E staining. (b) The expression of NLRP3, P62, LC3BII, LC3BI, and Wnt5a proteins in the lung tissues by Western blot. Three Western blots were replicated for the graph. The blots were cut prior to hybridisation with antibodies during blotting. (c) IL-1β level in the lavage fluid of mice by ELISA. (d) Immunofluorescence of M1 macrophage marker CD80 in mice lung tissues. * p < .05 vs. control group. # p < .05 vs. PM2.5 group. △p < .05 vs. PM2.5 + BOX5 group.
According to immunofluorescence, the intensified CD80 staining was occurred in the lung tissues of the PM2.5 group mice when compared to the control group. Oppositely, the attenuated CD80 staining was found in mice lung tissues of the PM2.5 + BOX5 group when compared to the PM2.5 group. Matched to the PM2.5 + BOX5 group, mice of the PM2.5 + BOX5 + Rapamycin group displayed the enhanced CD80 staining in the lung tissues (Figure 2(d)). Thus, all of these data implied that the inhibition of Wnt5a attenuated the PM2.5 induced lung injury in mice and the activation of autophagy counteracted this effect.
The morphology and elements of PM2.5 and its effect on MH-S cell viability
The morphology of PM2.5 was observed using the SEM. The size of PM2.5 was no more than 2.5 μm in the diameter. The morphology of PM2.5 was irregular, which exhibited as rod-like and granular shapes (Figure 3(a)). EDS was used for the elemental analysis of PM2.5. The element map of PM2.5 showed that, the elements included in PM2.5 contained carbon (C), oxygen (O), sodium (Na), magnesium (Mg), aluminum (Al), silicon (Si), Aurum (Au), sulfur (S), potassium (K), calcium (Ca), and barium (Ba) (Figure 3(b)). The effect of PM2.5 on the viability of MH-S cells was explored by CCK-8 assay. PM2.5 at a concentration of 12.5, 25, 50, and 100 μg/mL could not obviously affect the viability of MH-S cells. However, PM2.5 at a concentration of 200 μg/mL much reduced the viability of MH-S cells (Figure 3(c)). Thus, this study used 100 μg/mL of PM2.5 to treat MH-S cells in the following study. The morphology and elements of PM2.5 and its effect on MH-S cell viability. (a) The morphology of PM2.5 by SEM (b) The elements analysis of PM2.5 by EDS. (c) PM2.5 effect on the viability of MH-S cell by CCK-8 assay.
PM2.5 activated the NLRP3 inflammasome and exacerbated the IL-1β release in MH-S cells
As shown in Figure 4(a), prominently higher expression of NLRP3, pro-caspase-1, cleaved caspase-1, pro-IL-1β, and pro-IL-18 proteins were discovered in MH-S cells of the PM2.5 group when relative to the control group (p < .05). In comparison to the control group, the IL-1β level in MH-S cells of the PM2.5 group was much increased (p < .05) (Figure 4(b)). These data indicated that PM2.5 treatment activated the NLRP3 inflammasome and exacerbated the IL-1β release in MH-S cells. PM2.5 activated the NLRP3 inflammasome and exacerbated the IL-1β release of MH-S cells. (a) The expression of NLRP3, pro-caspase-1, cleaved caspase-1, pro-IL-1β, and pro-IL-18 proteins in MH-S cells by Western blot. Three Western blots were replicated for the graph. The blots were cut prior to hybridization with antibodies during blotting. (b) IL-1β level in MH-S cells by ELISA. * p < .05.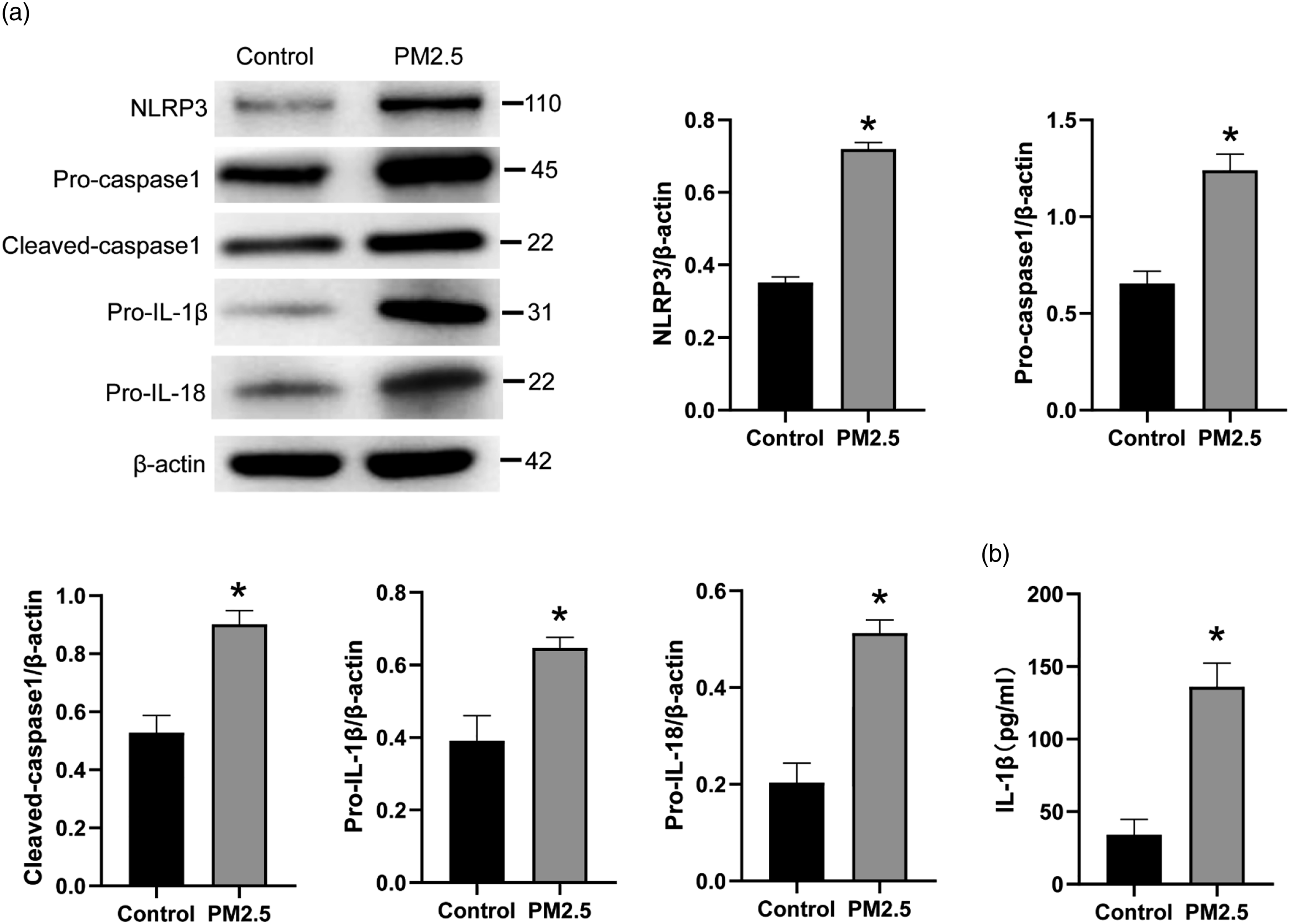
PM2.5 activated the autophagy and promoted expression of Wnt5a protein in MH-S cells
Western blot displayed a significantly higher expression of P62 and LC3BII/I proteins in MH-S cells of the PM2.5 group when compared with the control group (p < .05) (Figure 5(a)). Simultaneously, the expression of Wnt5a protein in MH-S cells of the PM2.5 group was higher than the control group (p < .05) (Figure 5(b)). Immunofluorescence exhibited that, the LC3B expression (red fluorescence) was higher in MH-S cells of the PM2.5 group than the control group (Figure 5(c)). From Figure 5(d), more LC3B-GFP positive autophagasome (green fluorescence) were found in MH-S cells of the PM2.5 group when matched to the control group. Thus, the autophagy and expression of Wnt5a protein in MH-S cells was activated by the PM2.5 treatment. PM2.5 activated the autophagy and promoted Wnt5a protein expression in MH-S cells. (a) The expression of LC3BI, LC3BII, and P62 proteins in MH-S cells by Western blot. Three Western blots were replicated for the graph. The blots were cut prior to hybridization with antibodies during blotting. (b) Wnt5a protein expression in MH-S cells by Western blot. Three Western blots were replicated for the graph. The blots were cut prior to hybridization with antibodies during blotting. (c) LC3B protein expression in MH-S cells by Immunofluorescence. (d) Autophagy assay of MH-S cells by LC3B-GFP punctae experiment. *p < .05.
PM2.5 activated the NLRP3 inflammasome and the IL-1β release in MH-S cells by facilitating the autophagy via activating Wnt5a
BOX5 (a specific inhibitor of Wnt5a) and Rapamycin (an autophagy activator) were used to treat MH-S cells. As shown in Figure 6(a), higher expression of Wnt5a, NLRP3, pro-caspase-1, cleaved caspase-1, pro-IL-1β, and pro-IL-18 proteins were observed in MH-S cells of the PM2.5 group, in comparison to the control group (p < .05). Interestingly, much lower expression of Wnt5a protein was found in MH-S cells of the PM2.5 + BOX5 group and the PM2.5 + BOX5 + Rapamycin group when matched to the PM2.5 group (p < .05). MH-S cells of the PM2.5 + BOX5 group had markedly lower expression of NLRP3, pro-caspase-1, cleaved caspase-1, pro-IL-1β, and pro-IL-18 proteins than the PM2.5 group (p < .05). However, in comparison to the PM2.5 + BOX5 group, the expression of NLRP3, pro-caspase-1, cleaved caspase-1, pro-IL-1β, and pro-IL-18 proteins were all significantly increased in MH-S cells of the PM2.5 + BOX5 + Rapamycin group (p < .05). PM2.5 activated the NLRP3 inflammasome and IL-1β release in MH-S cells by facilitating the autophagy via activating Wnt5a. (a) The expression of Wnt5a, NLRP3, pro-caspase-1, cleaved caspase-1, pro-IL-1β, and pro-IL-18 proteins in MH-S cells by Western blot. Three Western blots were replicated for the graph. The blots were cut prior to hybridization with antibodies during blotting. (b) IL-1β level in MH-S cells by ELISA. *p < .05 vs. control group. # p < .05 vs. PM2.5 group. △p < .05 vs. PM2.5 + BOX5 group.
ELISA presented remarkably higher IL-1β level in MH-S cells of the PM2.5 group when relative to the control group (p < .05). Oppositely, lower IL-1β level in MH-S cells of the PM2.5 + BOX5 group was found when compared with the PM2.5 group (p < .05). Relative to the PM2.5 + BOX5 group, the increased IL-1β level was observed in MH-S cells of the PM2.5 + BOX5 + Rapamycin group (p < .05) (Figure 6(b)). Hence, PM2.5 might activate the NLRP3 inflammasome and the IL-1β release in MH-S cells by facilitating the autophagy via activating Wnt5a.
PM2.5 facilitated the autophagy via activating Wnt5a in MH-S cells
According to Western blot, the expression of Wnt5a, P62, and LC3BII/I proteins was significantly increased in MH-S cells of the PM2.5 group when matched to the control group (p < .05). Lower Wnt5a protein was exhibited in MH-S cells of the PM2.5 + BOX5 group and the PM2.5 + BOX5 + Rapamycin group when compared to the PM2.5 group (p < .05). Intriguingly, remarkably lower expression of P62 and LC3BII/I proteins was observed in MH-S cells of the PM2.5 + BOX5 group when matched to the PM2.5 group (p < .05). Oppositely, the expression of P62 and LC3BII/I proteins was significantly elevated in MH-S cells of the PM2.5 + BOX5 + Rapamycin group when relative to the PM2.5 + BOX5 group (p < .05) (Figure 7(a)). LC3B expression in MH-S cells was detected using immunofluorescence. The enhanced LC3B staining (red fluorescence) was shown in MH-S cells of the PM2.5 group relative to the control group. Intriguingly, LC3B staining was attenuated in MH-S cells of the PM2.5 + BOX5 group than the PM2.5 group. However, matched to the PM2.5 + BOX5 group, MH-S cells of the PM2.5 + BOX5 + Rapamycin group exhibited the intensified LC3B staining (Figure 7(b)). Moreover, in comparison to the control group, MH-S cells of the PM2.5 group presented the enhanced LC3B-GFP positive autophagasome. However, less LC3B-GFP positive autophagasome were observed in MH-S cells of the PM2.5 + BOX5 group when relative to the PM2.5 group and the PM2.5 + BOX5 + Rapamycin group (Figure 7(c)). These results revealed that PM2.5 facilitated the autophagy in MH-S cells via activating Wnt5a. PM2.5 facilitated the autophagy via activating Wnt5a in MH-S cells. (a) The expression of Wnt5a, LC3BI, LC3BII, and P62 proteins in MH-S cells by Western blot. Three Western blots were replicated for the graph. The blots were cut prior to hybridization with antibodies during blotting. (b) LC3B protein expression in MH-S cells by Immunofluorescence. (c) Autophagy assay of MH-S cells by LC3B-GFP punctae experiment. *p < .05 vs. control group. # p < .05 vs. PM2.5 group. △p < .05 vs. PM2.5 + BOX5 group.
The ingestion of PM2.5 into MH-S cells
TEM was utilized to observe the ingestion of PM2.5 into MH-S cells. The images are shown in Figure 8. The red arrow represented autophagosomes, and the blue arrow represented PM2.5. MH-S cells of the control group had less autophagosomes than the PM2.5 group. Some PM2.5 particles were taken into MH-S cells of the PM2.5 group, the PM2.5 + BOX5 group and the PM2.5 + BOX5 + Rapamycin group. Less autophagosomes were observed in MH-S cells of the PM2.5 + BOX5 group when relative to the PM2.5 group. Matched to the PM2.5 + BOX5 group, MH-S cells of the PM2.5 + BOX5 + Rapamycin group had more autophagosomes. It is well known that the increased autophagosomes meant the enhancement of autophagy. Therefore, PM2.5 increased the autophagy in MH-S cells. However, Wnt5a inhibitor (BOX5) attenuated the PM2.5-induced autophagy in MH-S cells. Intriguingly, Rapamycin abrogated the effect of BOX5 on the PM2.5-induced autophagy in MH-S cells. The ingestion of PM2.5 into MH-S cells by transmission electron microscope.
Discussion
In recent decades, severe air pollution has posed a serious threat to the health of human, especially PM2.5 in the polluted air. In our previous study, PM2.5 has been detected to carry a large amount of harmful components, such as polycyclic aromatic hydrocarbons.
30
This study detected that PM2.5 contained several kinds of harmful elements, such as Au, Si, and S. The onset and development of many respiratory diseases has been reported to be related to PM2.5 in the atmosphere.31–33 In this study, PM2.5 exposure induced mice lung injury and activated the NLRP3 inflammasome, Wnt5a, and IL-1β release in mice lung tissues and MH-S cells. Interestingly, BOX5 treatment counteracted this effect of PM2.5. Moreover, Rapamycin abrogated the effect of BOX5 on the PM2.5-induced damage on mice lung tissues and MH-S cells. Thus, PM2.5 might activate the NLRP3 inflammasome and IL-1β release in MH-S cells by facilitating the autophagy via activating Wnt5a. The mechanism diagram is shown in Figure 9 (created by the authors). The mechanism diagram of PM2.5 induced lung injury.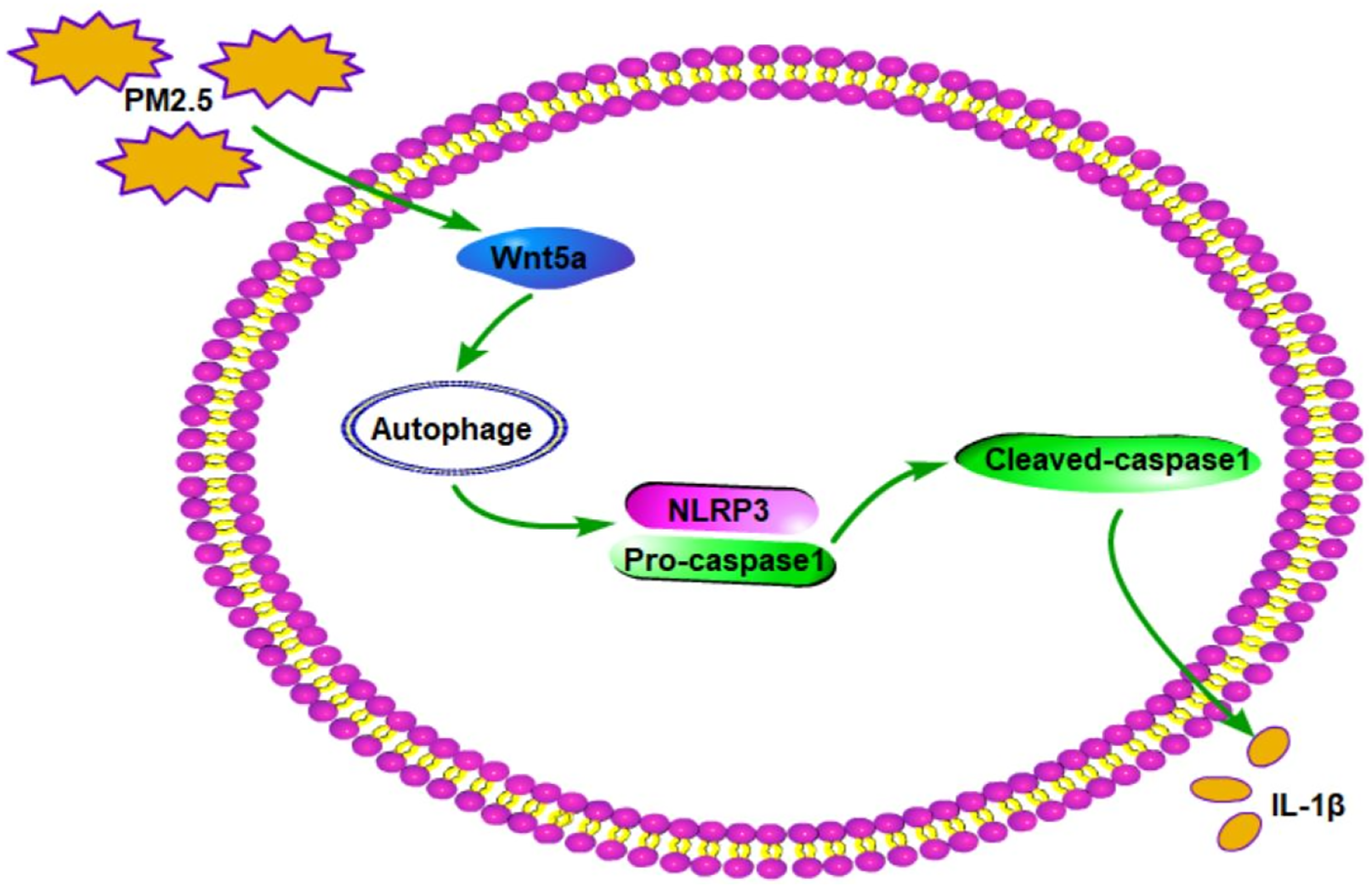
The innate immune system can respond initially to the irritants, and innate immune cells can detect pathogens with a fixed number of germline-encoded pattern recognition receptors. 34 Pattern recognition receptors detect the unique microbial structure called “pathogen-associated molecular pattern (PAMP)” Interestingly, the damaged cells can trigger the pattern recognition receptors through releasing the danger-associated molecular pattern (DAMP). After experiencing PAMP and DAMP, pattern recognition receptors can trigger the signaling cascades and then activate the nuclear factor-κB (NF-κB) to facilitate the formation of an inflammasome. 35 Pattern recognition receptor is a main component of inflammasome. 36 It can bind the nucleotide-binding oligomerization domain-like receptors (NLRs) to effector pro-caspase-1, thereby activating caspase-1. The activated caspase-1 further cleaves pro-IL-1β into IL-1β to induce the pro-inflammatory response.35,37 It has been identified that PM2.5 could activate the inflammasome through activating the NF-κB signaling. 36 The activation of the NLRP3 inflammasome is associated with the lung inflammation. 38 PM2.5 can promote the inflammatory response by activating the NLRP3 inflammasome.39,40 Recently, the activation of the NLRP3 inflammasome has been found to be involved in the pulmonary inflammation induced by PM2.53. More data about PM2.5 regulating the activation of NLRP3 to trigger lung inflammation are rarely to be found. In this study, PM2.5 exposure enhanced the expression of NLRP3, pro-caspase-1 and cleaved caspase-1 proteins and the release of IL-1β in mice lung tissues and MH-S cells. Typically, caspase-1 can be activated within the NLRP3 inflammasome, which further lead to the cleavage of pro-IL-1β into mature IL-1β. Thereafter, IL-1β releases to cells to cause the inflammation of cells and tissues. 41 NLRP3 and pro-caspase-1 proteins are two important markers of the activated NLRP3 inflammasome. 12 Pro-IL-18 is one of the major components of the NLRP3 inflammasome, which can cause the inflammatory damage to lung tissues. 42 This paper indicated that PM2.5 treatment induced the expression of pro-IL-18 in mice lung tissues. Moreover, it is well known that M1 macrophages exert a pro-inflammatory function through releasing the pro-inflammatory factors (such as IL-1β) and then exacerbate the inflammatory responses in lung tissues.43–45 In this paper, PM2.5 increased the expression of M1 macrophage marker CD80 in mice lung tissues, indicating the M1 polarization of macrophages triggered by the PM2.5 treatment. This study confirmed that PM2.5 induced the inflammation in mice lung tissues and MH-S cells by activating the NLRP3 inflammasome.
Autophagy is ubiquitous in eukaryotic cells, which is a highly conserved and homeostatic process. 46 Under normal circumstances, autophagy can maintain the stability of the intracellular environment by removing the damaged organelles, etc. 47 However, over activation of autophagy can induce the excessive degradation of cellular contents, thereby resulting in the impaired cell function and autophagic death. 48 Previous research has reported that autophagy was over-induced in the damaged lung tissues caused by PM2.5. 49 P62 is a classical receptor of autophagy. 50 In mouse model exposed to PM2.5, PM2.5 has been revealed to cause the lung tissue damage by triggering autophagy via increasing the expression of P62. 51 PM2.5 triggered the reproductive toxicity to male rats by inducing autophagy activation via promoting the expression of P62. 52 Similarly, this study indicated that PM2.5 exposure enhanced the expression of P62 protein in mice lung tissues and MH-S cells.
LC3B is the most important subtype of LC3, which is an important marker of autophagy.53,54 The elevated LC3BII/I ratio has become the main criterion for judging autophagy level. 55 A previous study has reported that PM2.5 treatment induced the activation of autophagy in A549 cells by increasing the expression of LC3BII. 29 In this paper, PM2.5 treatment promoted the expression of LC3B protein in MH-S cells and elevated the LC3BII/I ratio in mice lung tissues and MH-S cells. These data indicated that PM2.5 treatment activated the autophagy in mice lung tissues and MH-S cells. Previous study has reported that the NLRP3 inflammasome could be activated by autophagy. 56 Similarly, this study revealed that, PM2.5 could activate the NLRP3 inflammasome and IL-1β release in mice lung tissues and MH-S cells by facilitating the autophagy.
It has been found that PM2.5 increased the expression of Wnt5a in human bronchial epithelial cells. 23 However, the activation of Wnt5a could facilitate the inflammatory response in lung tissues. 57 To data, whether PM2.5 induces lung injury by activating Wnt5a has not been fully elucidated. This paper revealed that PM2.5 increased the expression of Wnt5a in mice lung tissues and MH-S cells. BOX5 (a specific antagonist of Wnt5a) treatment could reverse the promoting effect of PM2.5 on the lung injury, the NLRP3 inflammasome activation, IL-1β release, and autophagy in mice or MH-S cells. Thus, PM2.5 might activate the activation of the NLRP3 inflammasome and promoted the IL-1β release and autophagy in mice lung tissues and MH-S cells by activating Wnt5a. Moreover, Rapamycin (an autophagy activator) treatment reversed the inhibitory effect of BOX5 on the NLRP3 inflammasome activation, IL-1β release and autophagy in mice lung tissues and MH-S cells induced by PM2.5. Therefore, PM2.5 might activate the NLRP3 inflammasome and IL-1β release in MH-S cells by facilitating the autophagy via activating Wnt5a.
Of course, there were limitations in this study. First, macrophage recruitment or clearance in lung tissues upon PM2.5 treatment, PM2.5 + BOX treatment and PM2.5 + BOX + Rapamycin treatment should be investigated. Second, from Figure 1(a), (h), (e) staining indicated that PM2.5 treated mice exhibited more collagen components in the lung tissues. This suggests that PM2.5 treated mice might have lung fibrosis. Masson staining should be performed to verify this speculation. Moreover, a co-staining of macrophage maker and target proteins in mice lung tissues should be performed. Finally, sample size calculation should be done in this study. However, due to the laboratory limitations, these experiments cannot be performed at present. This issue will be focus of our future research.
Conclusion
PM2.5 treatment induced mice lung injury, increased the expression of Wnt5a, and activated the NLRP3 inflammasome, IL-1β release, and autophagy in mice lung tissues and MH-S cells. The inhibition of Wnt5a attenuated the lung injury of the PM2.5-induced mice. More importantly, PM2.5 might activate the NLRP3 inflammasome and IL-1β release in MH-S cells by facilitating the autophagy via activating Wnt5a. This paper provided a novel clue for pulmonary inflammation treatment induced by PM2.5. Clinically, pulmonary inflammation induced by PM2.5 could be treated by targeting the inhibition of Wnt5a. Of course, more research still needs to be done in the future.
Supplemental Material
Supplemental Material - PM2.5 activated NLRP3 inflammasome and IL-1β release in MH-S cells by facilitating autophagy via activating Wnt5a
Supplemental Material for PM2.5 activated NLRP3 inflammasome and IL-1β release in MH-S cells by facilitating autophagy via activating Wnt5aGuanli Yuan, Yinfeng Liu, Zheng Wang, Xiaotong Wang, Zhuoxiao Han, Xixin Yan and Aihong Meng in International Journal of Immunopathology and Pharmacologyr
Footnotes
Authors’ contributions
GY and AM conceived and designed the experiments. GY, YL, and XY performed the experiments. GY and ZW conducted the statistical analyses. GY, XW, and ZH wrote the paper. All authors read and approved the final manuscript submitted for publication.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Natural Science Foundation of Hebei Province (H2019206263), S&T Program of Hebei (15967753D, 19277760D) and Projects Funded by Hebei Provincial Finance Department in 2020.
Ethics approval and consent to participate
All experimental protocols of animal handling and sampling were approved by the Institutional Animal Care and Use Committee of China Medical University (No. CMU2021111), and all efforts were made to minimize the suffering of animals according to recommendations proposed by the European Commission (1997). The study was carried out in accordance with the approved protocol. All methods were conducted in accordance with relevant guidelines. All methods are reported in accordance with ARRIVE guidelines. Consent to participate: Not applicable.
Availability of data and material
All data generated or analyzed during this study are available from the corresponding author upon reasonable request.
Supplemental Material
Supplementary material for this article is available on the online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.