
Abstract
The development of regenerative therapies for central nervous system diseases can likely benefit from an understanding of the peripheral nervous system repair process, particularly in identifying potential gene pathways involved in human nerve repair. This study employed RNA sequencing (RNA-seq) technology to analyze the whole transcriptome profile of the human peripheral nerve in response to an injury. The distal sural nerve was exposed, completely transected, and a 1 to 2 cm section of nerve fascicles was collected for RNA-seq from six participants with Parkinson’s disease, ranging in age between 53 and 70 yr. Two weeks after the initial injury, another section of the nerve fascicles of the distal and pre-degenerated stump of the nerve was dissected and processed for RNA-seq studies. An initial analysis between the pre-lesion status and the postinjury gene expression revealed 3,641 genes that were significantly differentially expressed. In addition, the results support a clear transdifferentiation process that occurred by the end of the 2-wk postinjury. Gene ontology (GO) and hierarchical clustering were used to identify the major signaling pathways affected by the injury. In contrast to previous nonclinical studies, important changes were observed in molecular pathways related to antiapoptotic signaling, neurotrophic factor processes, cell motility, and immune cell chemotactic signaling. The results of our current study provide new insights regarding the essential interactions of different molecular pathways that drive neuronal repair and axonal regeneration in humans.
Introduction
In contrast to the central nervous system (CNS), the axons of the peripheral nervous system (PNS) are capable of regenerating after injury 1 and re-establishing functional connections with their distal target 2 . This regeneration is enabled by the interaction between the injured axons, the nerve extracellular matrix, immune cells such as macrophages, and Schwann cells 3,4 . These nerve components individually contribute to peripheral nerve repair and interact with each other to further support a functional regeneration 5 . Following an injury to a peripheral nerve, the distal stump undergoes a process known as Wallerian degeneration. Schwann cells, macrophages, and neutrophils are the key players in this process. Together they breakdown myelin debris and clear the severed axonal membrane and cytoskeleton 6 . Reprogrammed Schwann cells also communicate with the fibroblasts to synthesize major adhesion proteins and a new extracellular matrix, which helps in bridging the proximal and distal stumps of the injured nerve fibers (for review, see Jessen and Mirsky 8 ). Under favorable conditions, the proximal segment of the injured axon sprouts new processes that reassociate with a new phenotype of Schwann cells, optimized for repair, and regenerate through the extracellular matrix of the distal nerve. In this way, the regenerating peripheral nerve fibers re-establish communication with the original distal target and restore some degree of function.
Myelinating and nonmyelinating (Remak) subtypes of Schwann cells undergo cellular reprogramming into a “repair” profile in response to PNS injury 7 . Repair Schwann cells shed their myelin, become mobile, and secrete trophic factors and other signaling molecules to promote axonal regeneration 8 . In response to an injury, Schwann cells release soluble growth factors such as nerve growth factor, brain-derived neurotrophic factor, insulin-like growth factor-1, hepatocyte growth factor, vascular endothelial growth factor, neurotrophin-3, and glial cell line-derived neurotrophic factor (GDNF) 9 –12 . These growth factors play a crucial role in inducing and guiding the regenerating distal end of the injured peripheral nerve fibers 13 .
Regulation of gene expression may be a therapeutic target for promoting the successful recovery of peripheral nerve after injury. Identification of those genes and how they interact is crucial to exploring strategies that enhance the neural protection, regeneration, and repair processes. Promoting successful regeneration in the CNS has been difficult in neurodegenerative diseases, traumatic brain injury, stroke, epilepsy, and other neurological disorders. We believe that understanding the gene expression changes that drive effective neural repair within the PNS may also help in identifying new therapeutic targets or methods that could enhance CNS neural regeneration.
Whole transcriptome profiling of gene expression in response to peripheral nerve injury can now be feasibly studied using RNA sequencing (RNA-seq) technology. RNA-seq combines molecular biology approaches for RNA amplification with bioinformatics tools for measuring and validating large RNA-seq datasets. This technique allows for quantitative measurements of thousands of gene transcripts using small (10 to 30 mg or less) quantities of tissue. For a review of the applications of RNA-seq see Han et al 14 .
Our study was conducted in conjunction with an ongoing clinical trial (clinicaltrials.gov:%NCT#02369003), the “DBS Plus” trial, which involves grafting of autologous peripheral nerve fascicles in patients with Parkinson’s disease (PD) at the time of deep brain stimulation (DBS) surgery 15 , an approach we call DPS Plus. To our knowledge, our study is the first of its type studying and analyzing the human peripheral nerve repair genetics in response to a transection injury within the same human subjects. The central hypothesis of this ongoing clinical trial is that the progression of motor symptoms of PD may be slowed and modified by providing a source of neuroprotective and pro-regenerative factors, i.e., autologous peripheral nerve tissue, to the degenerating dopaminergic neurons responsible for the motor deficits of PD. The trial involved collection of two samples of the sural nerve from the same participant at two distinct time points. The first sample, referred to as the “pre-lesion” sample, was collected from the participant’s sural nerve during Stage I of the surgery, which involved DBS hardware implantation. The second sample, referred to as the “post-lesion” sample, was taken 2 weeks later from the distal end of the same nerve during Stage II of the surgery, which is the stage when the DBS leads are positioned into the subthalamic nucleus (STN) or globus pallidus internus (GPi). This two-stage approach corresponds with the two stages of DBS surgery and was designed with the objective of inducing pro-regenerative changes in the peripheral nerve following an injury 13,16 . This current study was performed to analyze the transcriptome profile of normal and transected sural nerve tissue in patients with PD and to help evaluate how a conditioning injury of peripheral nerve tissue can induce pro-regenerative changes.
Materials and Methods
Research Subjects
The nerve samples were collected from human subjects with PD who electively consented to participate in the clinical trial testing the safety and feasibility of peripheral nerve grafts to the brain for the treatment of PD. The consent allowed for the collection of sural nerve sample for research studies. The nerve samples of six participants (two females and four males), with a mean age of 64
Tissue Collection
The sural nerve samples were collected at two different time points: a pre-lesion sample was collected at the time of the first stage of DBS surgery (Stage I) followed by a post-lesion sample from the second stage of DBS surgery (Stage II) 2 weeks later 17,18 (Fig. 1). Both nerve samples were collected before activating the DBS electrodes following the second stage of DBS surgery. For the pre-lesion tissue collection, the surgeon made a transverse incision through the skin of the lateral ankle, 2 cm superior to the lateral malleolus. The surgeon then identified the neurovascular bundle containing the sural nerve with the associated artery and vein. The sural nerve was then separated from the bundle using blunt dissection. Next, two black silk sutures were tied around the nerve, roughly 1 cm apart, to mark the nerve. The section of the sural nerve between these sutures was then cut, a 1-cm nerve segment was removed, and the surgical area was sutured and closed.

Photographs of the peripheral nerve fascicles analyzed in this study. (A) The samples collected in Stage I of the DBS surgery (pre-lesion sample). (B) The samples collected in Stage II of the DBS surgery (post-lesion sample). DBS: deep brain stimulation.
The removed section of nerve was then stripped of its epineurium using microsurgical dissection in cold sterile saline. Individual fascicles of nerve fibers were separated using jeweler’s forceps, and the perineurium was discarded (Fig. 1). These fascicles were placed in conical microcentrifuge tubes, snap-frozen on dry ice, and stored at −80°C until they were assayed. For post-lesion tissue collection, the surgical area from Stage I was reopened at the original incision. The surgeon located the suture markers on the distal stump of the transected sural nerve and removed a new 1 to 2 cm segment of the remaining distal stump of the previously severed nerve. The tissue was kept on sterile saline ice and was prepared for the trial intervention as previously described 17 . Once the trial intervention was completed, the remaining fascicles (Fig. 1B) were placed in conical microcentrifuge tubes, snap-frozen on dry ice, and stored at −80°C until assayed.
RNA Extraction
RNA isolation was performed by homogenizing sural nerve fascicles in 1 ml of TRI Reagent Solution (ThermoFisher AM9738, USA) using a Fisher Scientific Power Gen 35 homogenizer (Fisher Scientific PG35-362, USA) with a microtip homogenizing probe. The homogenized lysate was transferred to a pre-pelleted 5Prime Phase Lock Gel—Heavy 2 ml tube (ThermoFisher NC1093153, USA) and incubated at room temperature for 5 min. Two hundred microliters of chloroform (Sigma-Aldrich C2432, USA) was added and the tube was shaken vigorously by hand for 15 s. Phase separation was performed by microcentrifugation at 12,000 × g for 10 min. The RNA containing aqueous phase was taken from the top of the Phase Lock Gel layer and transferred to a 1.7-ml microfuge tube. RNA was precipitated by adding 0.5 ml isopropyl alcohol {2-Propanol} (Sigma-Aldrich I9516, USA), mixed by repeated inversion, and incubated at room temperature for 10 min. RNA was pelleted by microcentrifugation at 12,000 × g for 10 min at 4°C. RNA pellet was washed two times with 80% ethanol (Sigma-Adrich E7023, USA) using a 7,500 × g microcentrifugation for 5 min at 4°C to pellet RNA between washes. RNA pellets were air dried 5 to 10 min at room temperature and resuspended in 25 µl of nuclease-free water. RNA purity was assessed by OD260/OD280 ratio calculation using a ThermoFisher NanoDrop 1000 Spectrophotometer (ThermoFisher ND-1000, USA). RNA Integrity was assessed by Agilent Bioanalyzer 2100 (Agilent G2939BA, USA) using the Eukaryotic Total RNA Nano assay.
RNA Sequencing
RNA-Seq was performed at a strand-specific 100 cycle paired-end resolution, in an illumina HiSeq 2500 sequencing machine (Illumina, San Diego, CA, USA). In a repeated measure design, mRNA from the six individual samples were sequenced pre- and post-lesioning, thus, resulting in a total of 12 samples. The 12 samples were multiplexed in two lanes of a flow cell, resulting between 25 and 34 million reads per sample. The read quality was assessed using the FastQC software 19 . On average, the per sequence quality score measured in the Phred quality scale was above 30 for all the samples. The reads were mapped to the human genome (GRCh38) using the STAR software, version 2.3.1z 20 . On average, 96.4% of the sequenced reads mapped to the genome, resulting between 24.3 and 32.8 million mapped reads per sample, of which on average 89% were uniquely mapped reads. Transcript abundance estimates were calculated using HTSeq (version 0.6.1) 21 . Expression normalization and differential gene expression calculations were performed in edgeR (release 2.14) 22 to identify statistically significant differentially expressed genes. A paired sample design was used in edgeR, which employs a negative binomial generalized linear model for statistical calculations. The edgeR package implements advanced empirical Bayes methods to estimate gene-specific biological variation under minimal levels of biological replication. The RNA composition in each sample was normalized in edgeR using the trimmed mean of M-values method. The significant P-values were adjusted for multiple hypotheses testing by the Benjamini and Hochberg method 23 as modified by Storey 24 , providing a false discovery rate q value for each differentially expressed gene. Genes with an absolute fold difference ≥2 and q ≤ 0.05 were considered statistically significant.
RNA-Seq Analysis
RNA-seq data were organized in a Microsoft Excel table for subsequent analyses. These normalized read counts (counts per million) were used to calculate fold change between the pre-lesion and post-lesion samples, and the log base 2 of fold change was used for further analysis.
Correlation matrices and heat maps, as well as volcano plots, were generated in Excel. Functional overrepresentation analysis on the GO set of term annotations 25 was performed using DAVID pathway analysis 26 . DAVID is an online software tool that groups genes based on their functional similarity. Given a list of significantly differentially expressed genes, DAVID uses a fuzzy clustering algorithm, that uses information in its knowledge base on genes and their functional associations, to group genes that are together, statistically significant in their association to a set of common functional categories 26 .
AmiGO GO annotations (http://www.geneontology.org/) for terms of interest were cross-referenced with significantly differentially expressed genes, yielding a list of differentially expressed genes related to each GO annotation’s respective biological function. The GO annotations chosen to be visualized in heat maps were selected based on their relevance to peripheral nerve repair 16 . Statistical criteria of q ≤ 0.05 and |FC| ≥ 2 were selected, yielding a total of 3,641 differentially expressed genes included in this analysis. Heat maps of the qualifying genes were generated using JMP Pro 13 (SAS). Hierarchical clustering was performed using Ward’s method in JMP.
Results
Tissue Samples
The mass and freezing delay time (time from when the nerve was removed from participant to when it was snap-frozen in dry ice) were calculated for the pre-lesion and post-lesion tissue (Table 1). Freezing time delay was significantly longer (paired two tailed t-test: t(5) = 4.863, P = 0.004) for post-lesion nerve samples (59 ± 25 min; mean ± SD) than pre-lesion ones (18 ± 5 min). This difference was a result of the longer surgical procedures needed for Stage II of the DBS surgery. The mitigation of Mass Effect during the RNA Access library preparation was accomplished in multiple ways through normalizations incorporated at multiple steps through the entire process of library preparation and sequencing.
Mass and Freezing Time Delay of the Nerve Samples Collected During Stage I vs Stage II.
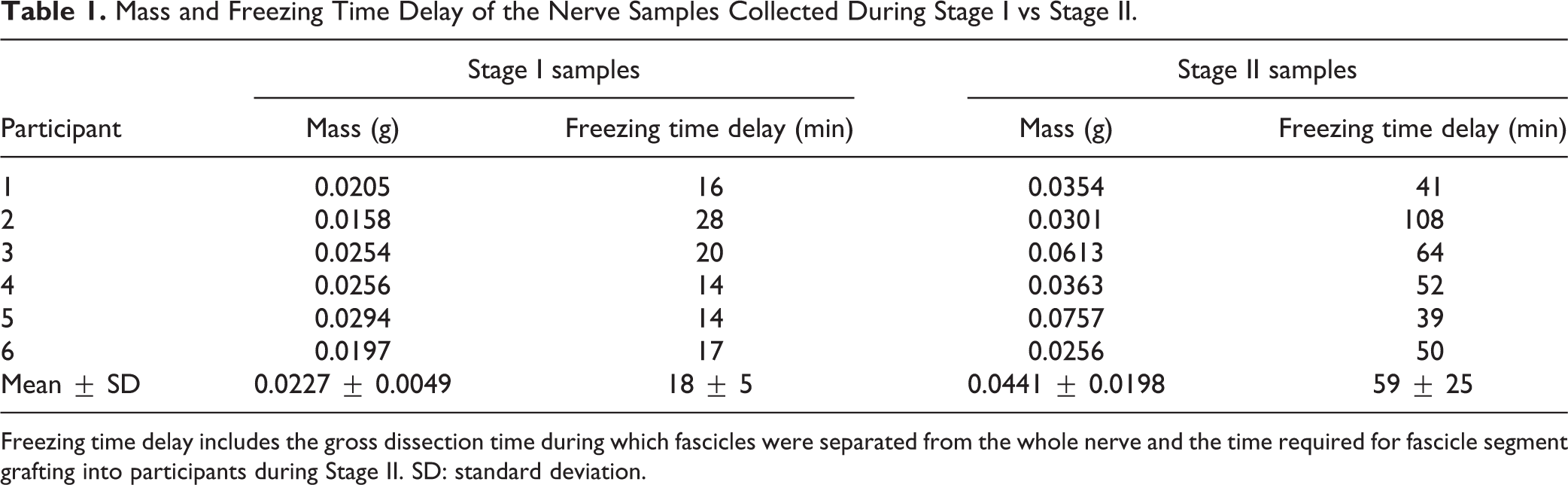
Freezing time delay includes the gross dissection time during which fascicles were separated from the whole nerve and the time required for fascicle segment grafting into participants during Stage II. SD: standard deviation.
Correlation Matrix and Volcano Plot
Analysis of the samples showed statistically significant changes in RNA levels among all six participants in response to the nerve transection lesion (Fig. 2A), with significant differences between the pre- and post-lesion samples and similarities within each of these groups as assessed by Pearson r correlation.
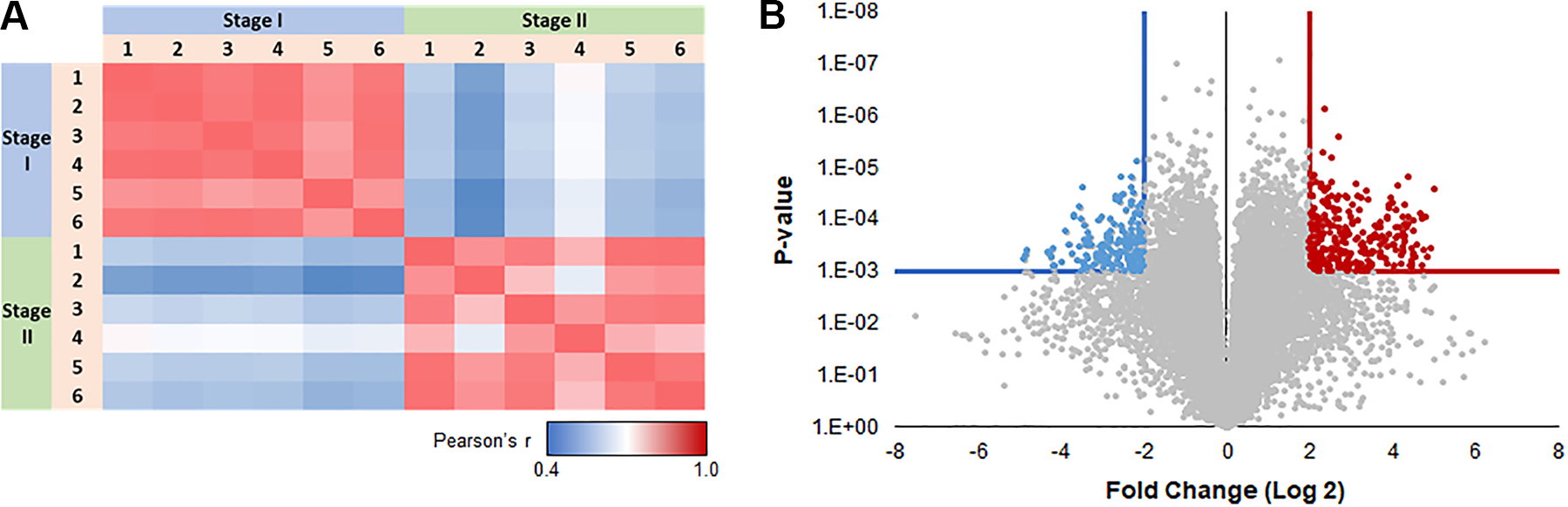
(A) Correlation matrix or Pearson’s r for the transcriptional profile of every subject vs every other subject. Scale bar: Correlation values range from 0.4 (blue—less similar) to 1 (red—more similar). This visualization shows strong agreement among different profiles within each stage, and a sharp distinction between stages. (B) Differences between Stage 1 and Stage 2. Log 2 scale fold changes (x-axis) are plotted as a function of P-value (inverted log 10 scale—volcano plot). Results that exceed conservative q-value (q ≤ 0.0003) and fold change (|FC| ≥ 4) cutoffs are highlighted (blue—downregulated in Stage 2, red—upregulated in Stage 2). FC: fold change.
The Volcano plot (Fig. 2B) estimates the most meaningful changes in the gene transcriptome, between pre- and post-lesion that exceeded a cutoff of q ≤ 0.0003 and fold change |FC| ≥ 4. Of those significantly differentially expressed genes, 693 gene transcripts (orange) were increased and 576 gene transcripts were decreased (blue). This analysis demonstrates that even with a very conservative criterion, there were hundreds of differentially expressed genes between the pre- and post-lesion tissue samples.
DAVID Pathway Analysis
DAVID pathway analysis revealed that the majority of the most significantly upregulated pathways (Table 2) were related to cell cycle, cell proliferation, and immune cell function. The majority of the most significantly downregulated pathways (Table 3) were related to synaptic structure and neuron function.
Top Significantly Increased Gene Ontology (GO) Pathways During Regeneration.

Top Significantly Decreased Gene Ontology (GO) Pathways During Regeneration.

Growth Factor Activity
Figure 3 shows all significantly differentially expressed (q < 0.05, |FC| > 2) gene transcripts annotated with the Gene Ontology (GO) term “Growth Factor Activity” (GO:0008083). Out of 166 unique genes with this GO annotation, 43 (25.9%) were significantly differentially expressed between pre- and post-lesion. Twenty-six of those genes were more abundant, whereas 17 genes were less abundant in post-lesion tissue in comparison with pre-lesioning levels.

Heat map showing all significantly differentially expressed (q < 0.05, |FC| > 2) gene transcripts annotated with the GO term “Growth factor Activity” (GO:0008083). Genes are organized by Ward hierarchical clustering. Dendrograms are scaled to hierarchical clustering distance; longer branches represent more distant clusters. FC: fold change; GO: gene ontology.
Myelination
Figure 4 shows all significantly differentially expressed (q < 0.05, |FC| > 2) gene transcripts annotated with the GO term “Myelination” (GO:0042552). Out of 110 unique genes with this GO annotation, 48 genes (43.6%) were significantly differentially expressed. Seven genes were more abundant and 41 genes were less abundant in response to the injury.

Heat map showing all significantly differentially expressed (q < 0.05, |FC| > 2) gene transcripts annotated with the GO term “Myelination” (GO:0042552). Genes are organized by Ward hierarchical clustering. Dendrograms are scaled to hierarchical clustering distance; longer branches represent more distant clusters. FC: fold change; GO: gene ontology.
Schwann Cell Transdifferentiation
Using the GO term “Epithelial–Mesenchymal Transition” (GO:0001837), 130 unique genes were yielded (Fig. 5). Of these, 29 genes (22.3%) were significantly differentially expressed (24 genes were more abundant and 5 genes were less abundant in post-lesion samples than pre-lesion samples). Likewise, the GO term “Schwann Cell Differentiation” (GO:0014037) identified 36 unique genes. Of these genes, 15 (41.7%) were significantly differentially expressed, with only 1 gene being more abundant while the other 14 genes less abundant in Stage II samples than Stage I samples.

Heat map showing all significantly differentially expressed (q < 0.05, |FC| > 2) gene transcripts annotated with the GO term “Epithelial–Mesenchymal Transition” (GO:0001837) or “Schwann Cell Differentiation” (GO:0014037). Genes are organized by Ward hierarchical clustering. Dendrograms are scaled to hierarchical clustering distance; longer branches represent more distant clusters. FC: fold change; GO: gene ontology.
Neuroprotection
“Negative Regulation of Apoptotic Processes” is defined by “Any process that stops, prevents, or reduces the frequency, rate or extent of cell death by apoptotic process.” And Negative Regulation of Neuron Death is defined as “any process that stops, prevents or reduces the frequency, rate or extent of neuron death.” Figure 6 shows all significantly differentially expressed gene transcripts annotated with the GO term “Negative Regulation of Apoptotic Processes” (GO:0043066) or “Negative Regulation of Neuron Death” (GO:1901215). This ontology yielded 916 unique genes, of which 128 (14.0%) were significantly differentially expressed (all 128 genes were more abundant in Stage II samples than in Stage I samples). Meanwhile, for Negative Regulation of Neuron Death, we found 67 genes out of 206 (32.5%) were significantly differentially expressed (45 genes more abundant and 22 genes less abundant in post-lesion samples than in pre-lesion samples). These gene changes indicated significant increases in transcripts of genes related to the reduction of neuron death.
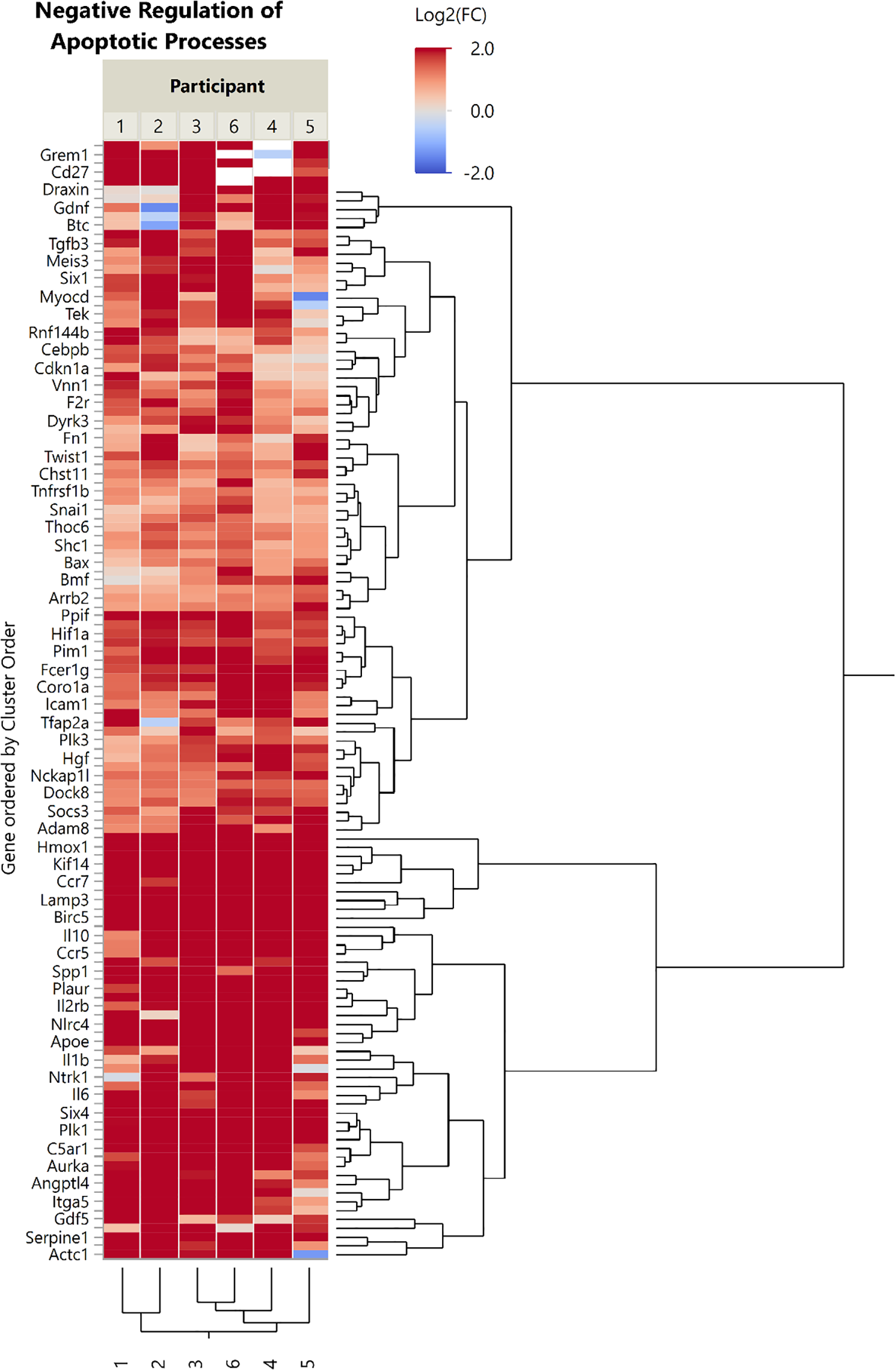
Heat map showing all significantly differentially expressed (q < 0.05, |FC| > 2) gene transcripts annotated with the GO term “Negative Regulation of Apoptotic Processes” (GO:0043066) or “Negative Regulation of Neuron Death (GO:1901215).” Genes are organized by Ward hierarchical clustering. Dendrograms are scaled to hierarchical clustering distance; longer branches represent more distant clusters. FC: fold change; GO: gene ontology.
Discussion
These results provide evidence that the transection lesion paradigm used in these surgeries induces phenotypic changes in the peripheral nerve tissue consistent with the peripheral nerve repair response: immune cell infiltration plus cell proliferation, Wallerian degeneration of axons, cessation of myelin synthesis, and upregulation of growth factors production 16,27,28 . DAVID pathway analysis revealed significant changes in genes related to the cell cycle, cell proliferation, immune cell function, synaptic structure, and neuron function. In addition, significant decreases in myelination-related gene transcripts were observed. The majority of significantly differentially expressed genes related to growth factor activity were upregulated, and of particular note was the expression of the GDNF gene. GDNF is known to promote neuroprotection and/or neurorestoration in the CNS and has been studied as a therapeutic for PD, reaching Phase I and II clinical trials in humans 29 –31 . These results also demonstrated a loss of markers of mature Schwann cell phenotype (MYOC 32 , FA2H 33 ) while those of the epithelial–mesenchymal transition increased (SNAI2 34 , WNT2 35 ). Taken together, these results indicate that the transection injury paradigm used in these human surgeries induced the transdifferentiation or reprogramming of peripheral nerve Schwann cells from a mature (myelinating) form into a repair (demyelinating) phenotype.
To better refine the results of our RNA-seq analysis to a more “biologically relevant” dataset, we decided to limit our observations to the genes whose transcript levels exceeded a fold change threshold of |FC| > 2. However, this convention may exclude genes that are biologically relevant at lower fold changes. For example, the transcription of NF2 gene, a marker of Schwann cell proliferation, was statistically significantly increased (P-value = 0.0192). However, the fold change of transcript levels was less than 2, so it was not included in the visualized data. Likewise, Mesencephalic Astrocyte Derived Neurotrophic Factor (MANF) transcripts level was significantly higher postinjury (P-value = 3.03E-07), yet its FC was 1.754 (data not shown). MANF plays a vital role in different reparative phases during the neuronal regeneration processes 36 , and MANF therapeutics are expected to enter clinical trials. Furthermore, we focused in this article only on transcripts that were differentially expressed between pre- and post-lesion samples while recognizing that some genes could be highly expressed in both stages, but not necessarily differentially. This is one limitation of this broad analysis approach, and in the specific case of NF2 levels in this tissue merits further study. The visualization of the negative regulation of apoptotic processes was also striking because over 100 genes were significantly differentially expressed and all were upregulated. This suggests that marked suppression of apoptosis was occurring in this tissue. Whether these antiapoptotic processes confer neuroprotection after tissue grafting merits further study.
The gene cluster of the growth factor terms showed multiple differentially expressed genes, with the majority being increased. One increased gene of note is GDNF, which is neuroprotective and neurorestorative of dopaminergic neurons and has been tried as a therapeutic intervention for PD 29,31,37,38 . All post-lesion nerve samples, except one, demonstrated an upregulation of GDNF transcription. Only the nerve sample collected from participant number 2 demonstrated a lower number of GDNF transcripts after injury (Fig. 3). The significant longer freezing time delay for this sample (Table 1) might have negatively affected GDNF-mRNA stability and its relevant count during RNA-seq processing. Multiple interleukins were also upregulated which, in addition to being cytokines, play a role in neurogenesis (for review, see Borsini et al 39 ). For example, the levels of gene transcript for IL-6, which has been described as neuroprotective against focal brain injury, were increased in response to the nerve injury 40 . The growth factor activity genes, which were decreased (e.g., CDNF) at 2 weeks postinjury are also of interest and may indicate the complexity of the neuronal repair process in regard to the changes of individual growth factors over time in response to nerve injury. That was evident in the work of Lin et al 41 as PPAR, PI3K-Akt, and chemokine signaling pathways were dominant in early Wallerian degeneration. Whereas at the later stage, the main signaling pathways were ErbB, tumor necrosis factor, AMPK, MAPK, PPAR, and Wnt 41 .
To our knowledge only Weiss and colleagues 42 have performed RNA-seq analysis on human peripheral nerve fascicles. They studied human peripheral nerve fascicles collected during surgeries, ex vivo degenerated nerves (for 8 d), cultured Schwann cells, and cultured fibroblasts. Their method of obtaining degenerated nerves differed from ours in that our model was degenerated in vivo and tissues were collected 2 weeks postinjury. Studies in animal models are more common. Arthur-Farraj et al 28 reported transcriptome and DNA methylome findings from mouse models. They found changes in epithelial–mesenchymal transition, which we also observed. Yi et al 43 studied a rat model of sciatic nerve crush injury and showed at acute, subacute, and post-acute time points the expression of inflammation and immune response; cellular movement, development, and morphology; and lipid metabolism, cytoskeleton reorganization, and nerve regeneration were all upregulated, respectively. Of note both Arthur-Farraj et al. and Yi et al. performed sequencing at multiple time points postinjury. This approach allowed them to show the change in differentially expressed pathways over time.
Myelination genes showed that significantly differentially expressed genes of this ontology were decreased 2 weeks after injury. This is consistent with myelin degradation after peripheral nerve injury as previously described 44 . In general, the Schwann cells showed overall decreased transcripts related to differentiation. This could be interpreted as transdifferentiation or reprogramming of Schwann cells 27 . However, this comparison was not straightforward. We expected to see that the JUN gene, which codes for the c-Jun transcription factor that regulates Schwann cell transdifferentiation, would be increased 27 . We found that the JUN gene was not significantly differentially expressed in these samples. We currently hypothesize that the time course of JUN transcription did not match with the two samples taken 2 weeks apart, as previous studies have identified increased JUN levels at 1 week post-lesion 28 . In fact, this study is limited overall in that it only addresses the levels of gene transcripts in the fascicles 2 weeks after the injury. Ideally, future studies should strive to investigate both shorter (3 to 7 days) and longer (3 to 6 weeks) postinjury periods to better understand the transdifferentiation process and the time course of the repair code.
The participants who donated tissue for this study had all been diagnosed with idiopathic PD and it is possible that the disease processes of this neurodegenerative disorder had some effect on peripheral nerve gene expression and its injury response, though this is not described in the available scientific literature. In other participants of the clinical trial (not included here), who have had a history of neuropathy, we have used clinical nerve conduction velocity tests to assess the sural nerve before grafting. None of those participants showed remarkable decrements in nerve conduction velocity (data not shown). A future study comparing baseline levels of RNA in patients with PD versus age-matched healthy controls could help determine potential baseline differences.
Because the underlying clinical trial necessitates a full transection of the sural nerve and excision of approximately 3 to 5 cm, participants commonly reported a painful sensation immediately postoperatively followed by numbness or tingling on the lateral aspect of the ankle and/or foot even after 1 yr. However, participants have not reported the sensation as painful or bothersome. The use of sural nerve excision in neurosurgical and plastic surgery applications has a robust literature and is shown to be well tolerated 45,46 .
With regard to the influence of variation of age and degree of PD within the study group, there was a remarkable consistency of transcriptome profiles within the pre- and post-lesion groups as assessed by the Pearson r correlation. The transcriptome profile of all participants in the pre-lesion group was similar, as was the transcriptome profile of all participants in the post-lesion group. The histology and transcriptome profiles of the peripheral nerve tissue in response to an injury were consistent with previous reports 28,42,43 . Furthermore, the correlation matrix visualizes the consistent RNA levels between participants as a result of the pre-lesioning approach 15,17 . These results support consistent transcriptome changes within the study group. Furthermore, these results support the feasibility and reproducibility of the time delayed approach for producing transdifferentiation of the sural nerve tissue into a repair cell phenotype in subjects with PD.
The freezing time delay difference between pre- and post-lesion samples is one of the limitations with the design of our study. This was inherent to the trial design where the focus was to optimize the surgical grafting procedure and the availability of tissue to be grafted during the DBS surgery. In Stage I the tissue was processed to isolate the fascicles immediately after extraction. During Stage II, the fascicles were removed from the sural nerve tissue, placed on saline ice, the sural nerve incision was closed, the graft locations in the brain were targeted, and the fascicle tissue was then grafted into the patient. We recognize that this delay was reflected in the difference in freezing time between the stages and it added to the variability between the samples. As a result, some of the transcriptome measurements might have been affected especially in the post-lesion samples compared to the in vivo state. Additionally, this analysis of peripheral nerve repair is in the context of a full transection injury. Other injury modalities, e.g., crush injuries, have been shown to induce different repair processes than transection injury 47 . One key difference is that while both crush and transection injury lead to Wallerian degeneration distal to the injury, in a crush injury the extracellular matrix scaffold remains intact, while this scaffold is interrupted in transection injury 48 . This should be considered when applying these findings to other injury models.
The sural nerve fascicles analyzed by RNA-seq included all cells and tissue components within the peripheral nerve. It should be noted that the tissue analyzed does not include the epineurium of the sural nerve. This was intentional, as it reflects the graft tissue composition (fascicles only), which is intended to contain mostly Schwann cells, macrophages, and extracellular matrix. This yielded an aggregate of all RNA in the tissue from multiple cell types. A future study using single-cell RNA-seq would be able to investigate the responses of individual peripheral nerve cells and cell types. Thus, it is possible that while we have emphasized the importance of the Schwann cells in the RNA-seq data and the “repair cell” properties of the tissue that the results cannot at this time be solely attributed to changes in Schwann cells. Furthermore, while Weiss et al. validated RNA-seq information using proteomics, this study focused on transcriptome changes in the peripheral nerve in response to injury but did not assess protein changes. Future studies could shed additional light as to the in vivo response of peripheral nerve in response to injury as well as the relationship of the transcriptome to the proteome of this dynamic tissue.
Taken together, the results of this study present whole-tissue transcriptome-scale data about the Wallerian degeneration affecting the distal stump of human peripheral nerve and at 2 weeks postinjury. The findings of this study reveal significant changes in the transcriptome of an injured human peripheral nerve after 2 weeks of repair processes in situ. Finally, this study provides data for future researchers to analyze and incorporate within their bioinformatics models of Wallerian degeneration. Such models may strengthen our understanding of the peripheral nerve repair process and its relevance to basic science as well as clinical and translational research that are looking to adapt peripheral nerve tissue/cells for use in promoting neuroprotection, neural repair, and regeneration for disorders of the CNS.
Supplemental Material
Supplemental Material, Gerhardt_RNASeq_spreadsheet - RNA Sequencing of Human Peripheral Nerve in Response to Injury: Distinctive Analysis of the Nerve Repair Pathways
Supplemental Material, Gerhardt_RNASeq_spreadsheet for RNA Sequencing of Human Peripheral Nerve in Response to Injury: Distinctive Analysis of the Nerve Repair Pathways by Andrew S. Welleford, Jorge E. Quintero, Nader El Seblani, Eric Blalock, Sumedha Gunewardena, Steven M. Shapiro, Sean M. Riordan, Peter Huettl, Zain Guduru, John A. Stanford, Craig G. van Horne and Greg A. Gerhardt in Cell Transplantation
Footnotes
Acknowledgments
Special thanks to Clark Bloomer and the University of Kansas Medical Center Genomics Core.
Ethical Approval
The study was approved by the University of Kentucky Institutional Review Board (#44749).
Statement of Human and Animal Rights
Procedures were conducted in accordance with the University of Kentucky Institutional Review Board’s (#44749) approved protocols.
Statement of Informed Consent
Written informed consent was obtained from participants for their anonymized information to be published.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Ann Hanley Parkinson’s Research Fund and the Clark Fund. The Genomics Core is supported by the following NIH grants—Kansas Intellectual and Developmental Disabilities Research Center (NIH U54 HD 090216), the Molecular Regulation of Cell Development and Differentiation—COBRE (P30 GM122731-03), the NIH S10 High-End Instrumentation Grant (NIH S10OD021743), and the Frontiers CTSA grant (UL1TR002366) at the University of Kansas Medical Center, Kansas City, KS, USA.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.