
Abstract
Myocardial infarction (MI) is the leading cause of morbidity and mortality in the world. The infarcted heart displays typical cell death cascades characterized by a loss of cells and fibrotic scarring in the myocardium. Cardiac hypertrophy and fibrosis largely contribute to ventricular wall thickening and stiffening, altogether defining an adverse cardiac remodeling that ultimately leads to impaired cardiac function and subsequent heart failure. Finding a strategy to promote therapeutic, instead of detrimental, cardiac remodeling may pose as a potent MI treatment. Accumulating evidence shows that microRNAs (miRNAs) may play an essential role in cardiovascular diseases. In particular, microRNA-133a (miR-133a) is one of the most abundant miRNAs in the heart. Multiple studies have demonstrated that miR-133a participates in the early pathology of MI, as well as in subsequent cardiac remodeling. In this review, we summarize recent research progress highlighting the regulatory effects of miR-133a in ischemic myocardial diseases, such as inhibiting angiogenesis, apoptosis, fibrosis, hypertrophy, and inflammation, while promoting therapeutic cardiac remodeling. The goal is to elicit a critical discussion on the translational direction of miRNA-mediated treatments towards a safe and effective MI therapy.
Overview of MicroRNA-133a (miR-133a) in the Heart
MicroRNAs (miRNAs) are endogenous, 19–22 nucleotide, and non-coding single-stranded RNA molecules that are important regulators of physiologic and pathologic conditions of the body. MiRNAs usually play critical roles in regulating a range of cellular processes by post-transcriptional suppression of their target genes. Certain miRNAs may be expressed in a tissue-specific pattern, such as cardiac miRNAs (miR-1, miR133a, miR-208a/b, and miR-499), which are abundantly expressed in the myocardium. There is mounting evidence that these miRNAs are involved in heart development and certain cardiovascular diseases, including myocardial infarction (MI), in both experimental animals and patients (Table 1) 1 . MiR-133 is transcribed from the same chromosomal loci as miR-1, and miR-1 has expression levels during cardiac hypertrophy that change in the opposite direction during myocardial infarction 1,4 . Of note, miR-133a is not only associated with heart development and disease, but is also involved in various cancers such as breast cancer and hepatocellular carcinoma 21,22 . In addition, miR-1 and miR-133a play a key role in promoting cardiogenesis, heart function, and pathology. While miR-1 and miR-133a predominantly control the early stages of cardiogenesis by directing the commitment of embryonic stem cells and mesodermal precursors to the cardiac-specific muscle lineage, in the heart, miR-1 and miR-133a also mediate cardiac conductance and automaticity by regulating all phases of the cardiac action potential 1 . Particularly, miR-133a is essential for proper heart development as deletion of both miR-133a genes leads to anomalous heart smooth muscle gene expression, deviant apoptosis and proliferation patterns, lethal ventricular septal defects, and disorganized sarcomeres 23 . It is also interesting to note that miR-133a and miR-1 may have opposite roles during cardiac differentiation 24 , and that miR-133 inhibits apoptosis while miR-1 promotes apoptosis and oxidative stress in damaged cardiomyocytes 1 –3 . Patients with MI may develop ventricular fibrillation (VF), a vital cause of death, and down-regulating miR-133a/b may contribute to the development of VF in patients with MI 14 . Furthermore, over-expression of miR-133 improves cardiac function in a rat model of MI such as by increasing the left ventricular ejection fraction (LVEF) and fractional shortening (FS) 25 .
Role/Expression of MicroRNAs in the Heart.

MiR-133a as a Potential Diagnostic Biomarker of Acute MI
MiR-133a is down-regulated in both the infarcted region and the border zone of the heart in MI patients, as well as in experimental animals 14,15 . In contrast, the expression level of miR-133a in serum is elevated significantly in patients with acute myocardial infarction (AMI) or with unstable angina pectoris 5,16,17 . Additionally, miR-133a levels in serum are significantly related to all-cause mortality in acute coronary syndrome (ACS) patients 16 . In 2011, Kimura et al. first measured the levels of circulating miR-1 and miR-133a associated with cardiovascular diseases, and demonstrated that the levels of circulating miR-1 and miR-133a are elevated early after the onset of chest pain when there is no up-regulation in serum creatine phosphokinase (CK or CPK) or cardiac Troponin T (cTnT), and that serum miR-133a levels are sensitive to myocardial injury compared with miR-1 levels 5 . Increased levels of circulating miR-133a are found in exosomes, which implies that the living myocardium may be the source of circulating miR-133a 5 . Moreover, elevated levels of circulating miR-133a are strongly associated with AMI diagnosis. In addition to traditional markers for clinical prognosis in AMI patients, the concentration of miR-133a may also provide prognostic information, perhaps even earlier than these traditional markers 5,26 . Indeed, based on their tissue specificity, cardiomyocyte-enriched miRNAs such as miR-1, miR208a, miR-208b, miR-133a, miR-133b, and miR-499 have been proposed as potential diagnostic markers in patients with AMI 16,27 –29 . Nevertheless, circulating levels of miR-133a and miR-423-5p have failed as useful biomarkers of left ventricular (LV) remodeling after MI 30 .
MiR-133a Modulating Angiogenesis Act as Angio-miR
The sprouting of new blood vessels by angiogenesis is key in physiologic vascular development and pathological homeostasis. Abnormal angiogenesis leads to severe pathological conditions such as ischemia and cancer. Recently, miRNAs have been implicated to be involved in certain angiogenic factors and signaling pathways, and use small non-coding RNAs to promote or suppress angiogenic processes. For instance, the up-regulation of miR-133a induced by diabetes mellitus impairs angiogenesis in peripheral arterial disease (PAD) by reducing NO in endothelial cells 31 . Additionally, miR-133a suppresses angiogenesis of endothelial cells including proliferation rate, cell viability, and migration activity via targeting of VEGFR2 and FGFR1 32 . MiR-133a is reduced when vascular smooth muscle cells (VSMCs) are inclined to proliferate in vitro and following vascular injury in vivo, and increase when VSMCs are coaxed back to quiescence in vitro and in vivo. In addition, miR-133a interference and over-expression experiments show that miR-133a plays a mechanistic role in VSMC proliferation. Among the possible targets of miR-133a, the most reliable is the serum response factor (SRF), which plays a critical role in muscle proliferation and differentiation depending on its association with co-factors such as myocardin, HOP, and Elk-1 33 –37 . Accordingly, adeno-miR-133a suppresses while anti-miR-133a improves VSMC proliferation and migration in vitro and in vivo, by suppressing the expression of the transcription factor, Sp-1 38 .
MiR-133a Reduces Hypoxia-Induced Apoptosis in Cardiac Myocytes
Apoptosis, also called programmed cell death, plays a key role in both the physical development and the pathology of a variety of cells and tissues. Myocardial hypoxia is a major cause of cardiac dysfunction as it triggers cell injury and apoptosis 39 . Growing evidence indicates that miRNAs are of vital importance in regulating cardiovascular diseases, necessitating the demonstration of the molecular mechanism by which miRNAs control apoptosis and the identification of their direct and indirect targets 40 –43 . MiR-133a is among the most abundant of the miRNAs present in the normal heart, and significant changes in the expression of miR-133a are observed in response to anoxia stress 44 . Interestingly, miR-133a is significantly down-regulated in hypoxic H9c2 cells, a type of SD rat cardiomyocyte, and the over-expression of miR-133a suppresses hypoxia-induced apoptosis and enhances cardiomyocyte survival 6,45 –47 . Additionally, miR-133a is an apoptosis suppressor in myocardial ischemic postconditioning (IPost), inhibiting TAGLN2, HSP60, HSP70, Apaf-1, caspase-3/8/9 expression, and promoting anti-apoptotic protein Bcl-2 expression 6,7 –9 . Ischemia and reperfusion injury (I/R injury) increases apoptosis via elevated expression of pro-apoptotic genes like caspase-9, and reduces miR-1 and miR-133 levels. In contrast, IPost up-regulates miR-133a, which decreases caspase-9 expression, and, consequently, decreases apoptosis of cardiomyocytes under I/R injury. Thus, myo-miRNAs miR-1 and miR-133a may play an important role in IPost protection by regulating apoptosis-related genes, such as caspase-9 7 .
MiR-133a Over-Expression Protects Against Cardiac Fibrosis Post-MI
The main determinants of tissue fibrosis are an activated transforming growth factor-β (TGF-β) signaling cascade and the accumulation of increased extracellular matrix (ECM) proteins such as fibronectin (FN1) and collagen 1 alpha 1 V (COL4α1) 48 . MiR-133a, along with other transcription factors or miRNAs, can induce myocardial transdifferentiation of cardiac fibroblasts by inhibiting TGF-β signaling or the expression of certain factors that promote fibrosis, such as snail-1 expression 49 –51 , as well as improve cardiac function and fibrosis by inhibiting Akt in heart failure 10 . Connective tissue growth factor (CTGF) is another important target of miR-133a and a key molecule in the process of fibrosis. Thus, CTGF appears to be a potential therapeutic target in MI, as miR-133a can potentially protect against cardiac fibrosis by decreasing CTGF expression in the heart 52 . Moreover, the SRF/CTGF/miR-133a axis plays an important role in regulating cardiac fibrosis 53 and miR-133a could be a potential therapeutic target for diabetes-induced cardiac fibrosis and related cardiac dysfunction 48 .
MiR-133a Represses Cardiac Hypertrophy
MiR-133a has potential regulatory roles in cardiac hypertrophy. Activation of NFAT (nuclear factor of activated T cells)-mediated hypertrophic signaling is a key regulatory response to hypertrophic stimuli. NFATc4, a hypertrophy-associated mediator, is a negatively regulated target of miR-133a 54 . Additionally, in vitro over-expression of miR-133 or miR-1 could inhibit cardiac hypertrophy. In contrast, inhibition of miR-133 by ‘decoy’ sequences induces hypertrophy, which is more pronounced than hypertrophy generated with common inducers. In vivo inhibition of miR-133 by a single transfection of an antagomir causes sustained and marked cardiac hypertrophy. RhoA, a GDP-GTP exchange protein regulating cardiac hypertrophy; Cdc42, a signal transduction kinase involved in hypertrophy; and Nelf-A/WHSC2, a nuclear factor implicated in cardiogenesis, have been identified as specific targets of miR-133 11 . In addition to MI, circulating miR-133a could also serve as a biomarker for predicting cardiac hypertrophy in chronic hemodialysis patients and after valve replacement surgery in patients with aortic stenosis 12,13 .
MiR-133a Promotes Regeneration and Cardiac Programming Post-MI
Regeneration of the infarcted heart with new, functional cardiomyocytes remains challenging, but promising. Transplantation of cardiac stem cells (CSCs) or progenitor cells has been regarded as a potential therapeutic option for myocardial infarction patients. At present, cell therapy approaches with various types of mature or stem cell patients have produced modest improvements. Among them, resident CSCs/CPCs are a promising option. The great beneficial cardiac-specific effect of cardiac miRNAs including miR-133a is very helpful for enhancing the regenerative properties and survival of transplanted stem cells and cardiac progenitor cells, and for reprogramming mature non-cardiac cells to cardiomyocytes 1 . Both miR-1 and miR-133a progressively increase early during in vitro cardiac differentiation of adult CPCs, but only miR-133a expression increases under in vitro oxidative stress. miR-1 promotes differentiation of CPCs, while over-expressed miR-133a protects CPCs from cell death by targeting the pro-apoptotic genes Bim and Bmf 51 . As a result, miR-133a-CPCs improve cardiomyocyte proliferation post-MI. The beneficial effects of miR-133a-CPCs seem to correlate with the enhanced expression of various related paracrine factors and the valid cooperative secretion of miR-133a via exosomal transport 51 . Direct reprogramming refers to changing mature cells from one lineage to another without passing through a stem cell state, and cardiac reprogramming indicates the conversion of other cells into cardiomyocytes. A combination of cardiac-miRNA-1, 133, 208, and 499 are capable of promoting the direct cardiac reprogramming of fibroblasts to cardiomyocyte-like cells in vitro 55 . Therein, miR-133 promotes cardiac reprogramming by targeting Snai-1 and silencing fibroblast signatures 50 . Growing evidence demonstrates that muscle specific miRNAs, also defined as myo-miRs, function as a control center in directing diverse biological processes during myogenic proliferation and differentiation 56 , and the most widely studied ones are members of the miR-1, miR-206, and miR-133 families 33,57 . Additionally, the myogenic transcription factors myogenin and myogenic differentiation 1 (MyoD) bind to regions upstream of the miR-1 and miR-133 stem loop, thereby providing a molecular explanation for the observed induction during myogenesis. An increase in the levels of miR-1, miR-133, and miR-206 is seen during myogenesis 58 , indicating miR-133’s role in the process.
The Role of miR-133a in Anti-Inflammation Post-MI
Endomyocardial miR-133a levels correlate with myocardial inflammation, such as macrophage infiltration 59 . Over-expressing miR-133a reduces inflammatory cell infiltration in the heart at 7 and 28 days post-MI 25 . In contrast, some clinical investigations show that serum miR-133a levels are significantly increased in critical illness and sepsis, are correlated with the severity of the disease, and predict an unfavorable outcome for critically ill patients 60 . Indeed, miR-133a may also serve as a pro-inflammatory miRNA, warranting more direct studies to confirm or disprove this conclusion.
MiR-133a and Stem Cell Transplantation in MI
Of note, transplanting stem cells into hearts afflicted with MI may improve the outcome of the condition. Indeed, bone marrow mesenchymal stem cells (MSCs) grafted into the MI heart generate healthy cardiomyocytes, increase cardiac function, promote angiogenesis, and decrease detrimental remodeling 61,62 . However, ischemic conditions in the infarcted heart hinder the survival of the transplanted stem cells and reduce these beneficial effects. MiRNAs play important roles in regulating cell apoptosis, differentiation, and proliferation and appear promising in bolstering the survival and efficacy of these transplanted stem cells 9 . In particular, miR-133a protects CPCs against apoptosis 51 , increases the proliferation of myoblasts 33 , and curtails cardiac remodeling, hypertrophy, and fibrosis 9 . The presence or absence of certain miRNAs such as miR-27b enables senescence to be modulated in MSCs 59 , and miRNAs such as miR-21 are critical for facilitating MSC-induced protection of human bronchial epithelial cells under hypoxic conditions 63 . Moreover, let-7-5p miR appears to be critical for enabling extracellular vesicles derived from Wharton’s jelly MSCs to prevent the apoptosis of neurons under perinatal hypoxia-ischemia 64 . Interestingly, transplantation of MSCs transfected with miR-133a in an MI heart enhances graft survival and cardiac function, and reduces cardiac fibrosis compared with transplantation with non-transfected stem cells 9 (Fig. 1). Additionally, in a rat myocardial infarct model, injecting MSCs transfected with the miR-133a agomir increases cardiac function and decreases MSC apoptosis, inflammation, and infarct size, relative to injecting MSCs alone. In contrast, injecting MSCs transfected with the miR-133a antagomir increases MSC apoptosis 25 . Furthermore, overexpression of miR-133a promotes the differentiation of human bone marrow-derived MSCs to cardiac cells that express markers such as cTnT and β-MHC, and executes these changes by targeting epidermal growth factor receptor 65 . Thus, miR-133a can be utilized in stem cell therapies for MI to augment the survival of grafted cells and increase treatment effectiveness, possibly by enabling more transplanted stem cells to differentiate into healthy cardiomyocytes, or by contributing additional cardioprotective benefits. Similarly, the survival rate and ability of transplanted cells to resist host-mediated immune responses may also be strengthened by employing exosomes derived from MSCs that overexpress indoleamine 2,3-dioxygenase 1 66,67 .
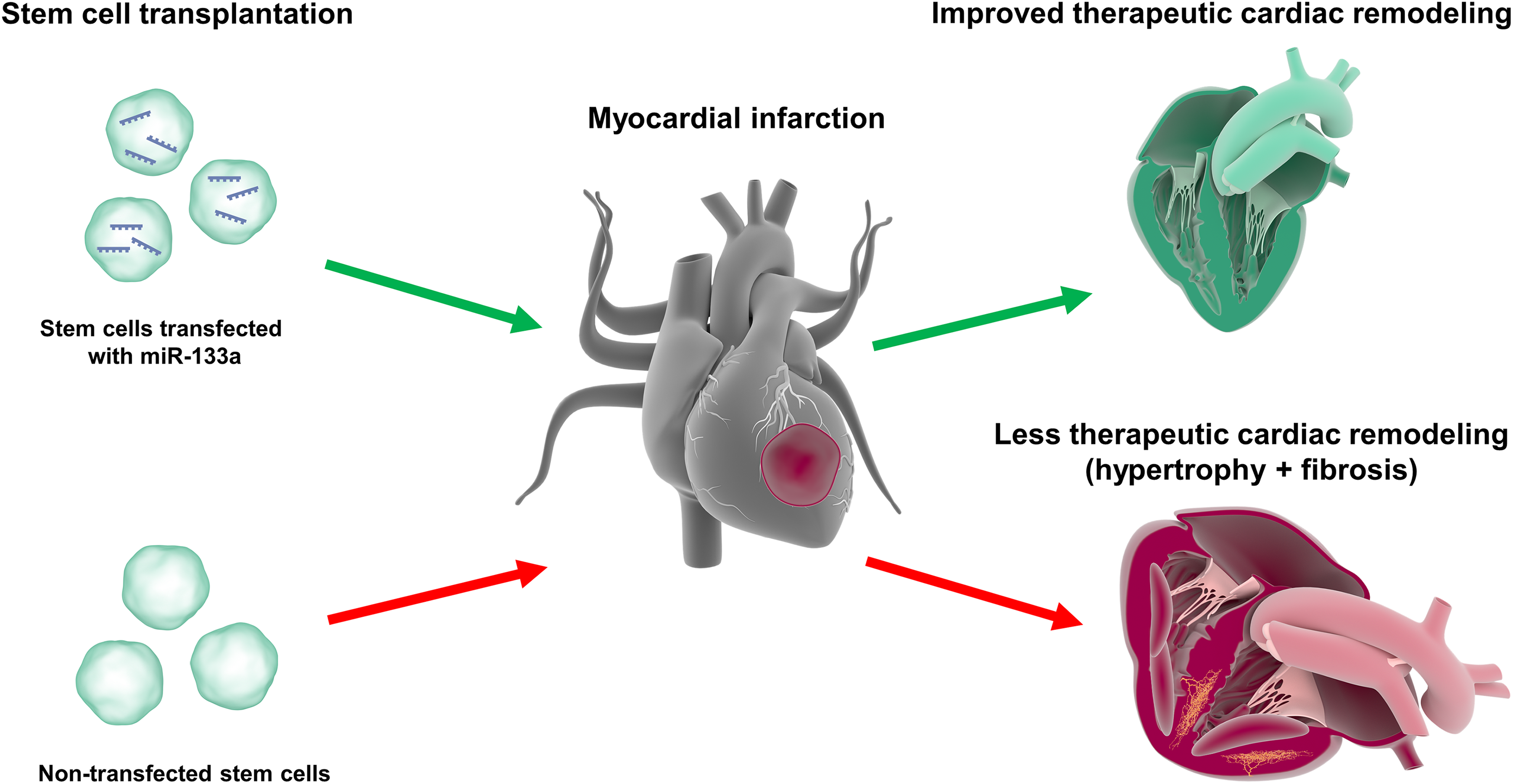
Therapeutic cardiac remodeling via transplantation of microRNA-133a (miR-133a) and stem cells. Myocardial infarction (MI) can lead to adverse cardiac remodeling and promote the development of hypertrophy and fibrosis. Transplantation of stem cells transfected with miR-133a in MI hearts can promote therapeutic cardiac remodeling to combat these detrimental effects and may be more effective than transplantation of stem cells alone.
Conclusion and Perspectives
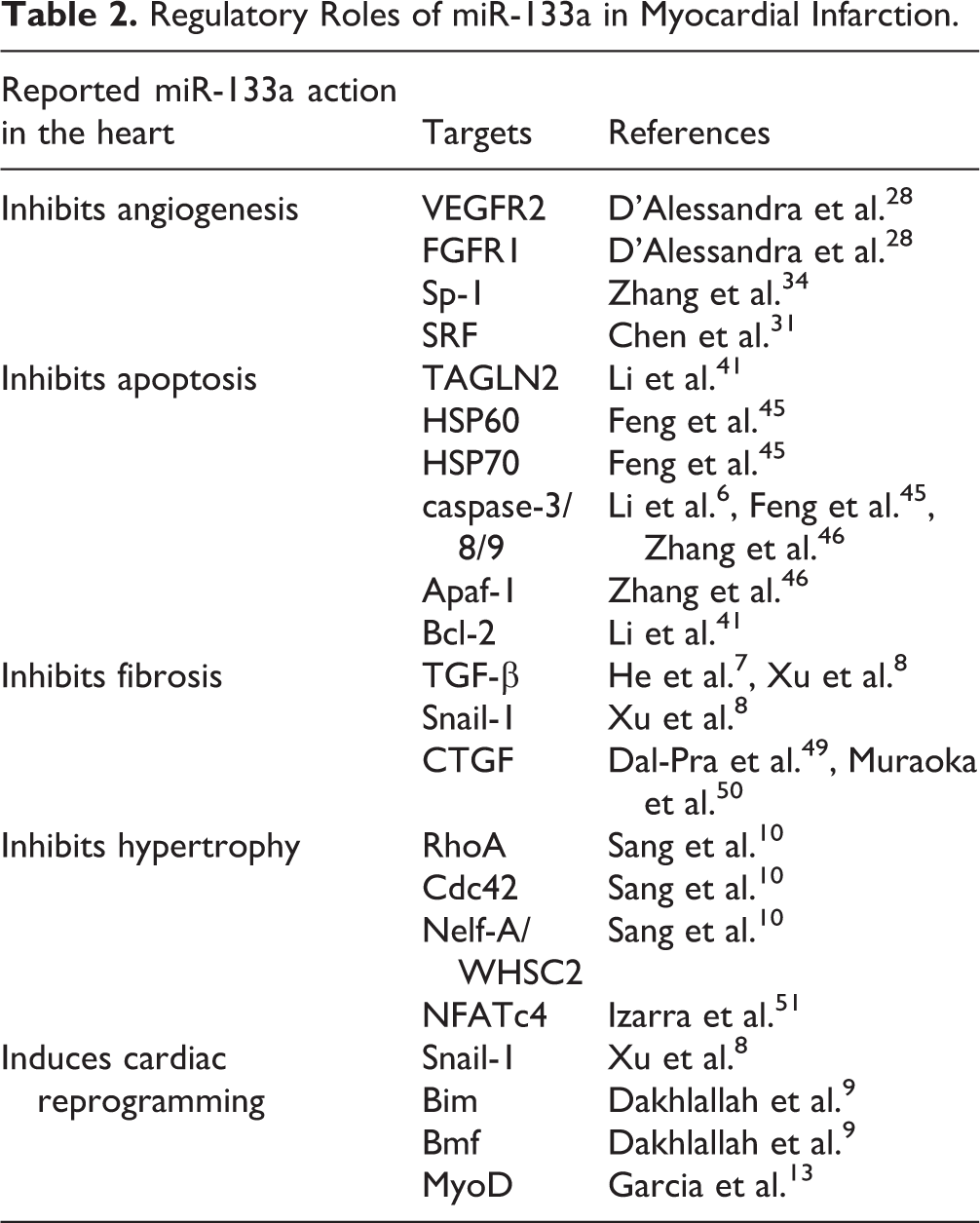
Collectively, these lines of evidence imply the potential of miR-133a as a promising therapeutic target in the treatment of MI. Nevertheless, the roles of miR-133a in myocardial infarction remain largely unknown. So far, studies have demonstrated that miR-133a induces positive effects on infarcted hearts in regard to angiogenesis, inflammation, apoptosis, fibrosis, hypertrophy, and cardiac programming (Table 2). However, we cannot conclude whether miR-133a is completely beneficial or not to the infarcted heart. The mechanisms of miR-133a-mediated cardiomyopathy are extremely complex because miR-133a targets the upstream molecules of various critical transcription factors. Moreover, numerous studies have shown that infarcted myocardium experience decreased miR-133a compared with normal areas, and up-regulating miR-133a expression through genetic manipulation techniques are beneficial for the heart under ischemic injury. These findings indicate that certain levels of miR-133a may be of vital importance in maintaining the balance of cardiac function. Therefore, the main aim of miR-133a intervention is to restore miR-133a levels in the myocardium, which is influenced by cardiac stress. In addition to the intracellular function of miR-133a, increasing evidence has revealed that circulating miR-133a can be used as a potential MI biomarker. Thus, it may be worthwhile to consider that miR-133a, in addition to being potential therapeutic target, can also be used as a diagnostic biomarker.
Regulatory Roles of miR-133a in Myocardial Infarction.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.