
Abstract
Induced pluripotent stem cells (iPSCs), which are generated through reprogramming adult somatic cells by expressing specific transcription factors, can differentiate into derivatives of the three embryonic germ layers and accelerate rapid advances in stem cell research. Neurological diseases such as amyotrophic lateral sclerosis (ALS) have benefited enormously from iPSC technology. This approach can be particularly important for creating iPSCs from patients with familial or sporadic forms of ALS. Motor neurons differentiated from the ALS-patient-derived iPSC can help to determine the relationship between cellular phenotype and genotype. Patient-derived iPSCs facilitate the development of new drugs and/or drug screening for ALS treatment and allow the exploration of the possible mechanism of ALS disease. In this article, we reviewed ALS-patient-specific iPSCs with various genetic mutations, progress in drug development for ALS disease, functional assays showing the differentiation of iPSCs into mature motor neurons, and promising biomarkers in ALS patients for the evaluation of drug candidates.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a fatal disease with a survival time of less than 5 years and prevalence of 3–4 per 100,000 persons in the United States 1,2 . The neurodegenerative disorder was first described in the late 19th century and is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, leading to paralysis 3,4 . Although ALS has been studied for many decades, scientific and clinical results have only partially revealed that genetic mutation may play a role in the onset of disease 5 –15 . In addition to hereditary factors, an unregulated neural transmitter system and environmental factors are the major factors influencing the progression of ALS 16 . These broad and general causes have confounded the investigation of therapies.
Although most cases of ALS are the sporadic form (sALS) of unknown etiology, ∼25% of ALS cases are the familial form (fALS), which is clinically and pathologically indistinguishable from sALS 17 . More detailed studies have been conducted on fALS than on sALS. Dominantly inherited gene mutations associated with fALS such as cytosolic copper-zinc superoxide dismutase 1 (SOD1), hexanucleotide repeat (chromosome 9 open reading frame 72, C9ORF72), tar DNA-binding protein 43 (TDP-43), fused in sarcoma (FUS), and other less frequent genes contribute to considerable genetic heterogeneity 6,17 –23 . Among the ALS-related gene mutations, SOD1 mutants are responsible for 20% of fALS 24 and the synergistic effect of the mutant Alsin (also known as ALS2) and SOD1 are linked to the early onset of ALS 7,25 . Genetically engineered cell lines or animal models for these genetic mutations are used to investigate the development of new drugs and to verify drug efficacy.
ALS modeling rodents or embryonic stem cells (ESCs) expressing ALS-causing genes have been generated to represent a specific subset of the syndrome in an effort to understand the onset and progression of ALS disease 22,26 –32 . However, the transgene method alone cannot be used for mimicking the real physiology of motor neurons in sALS patients because it cannot completely correlate the genotype with the neuronal phenotype 26,33 . Because animal and stem cell models for elucidating ALS pathogenesis have demonstrated shortcomings in recapitulating sALS patients, their capacity and application for the evaluation of therapeutic efficacy remain unmet needs. As an alternative, researchers have obtained induced pluripotent stem cells (iPSCs) from patients with neurodegenerative diseases, which have allowed the study of these cells with specific cell differentiation 2,34 –36 .
Human iPSCs can be produced by reprogramming the somatic cells for a state of embryonic-like stem cell via delivering the required transcription factors (Fig. 1A and B) 37 –39 . Such iPSCs can be directed to generate different kinds of neural cell types. iPSCs obtained from patients with ALS symptoms can be investigated at the molecular level to probe the changes in degeneration/death of motor neurons that trigger destruction of the neuromuscular junction 40 –42 . Therefore, differentiated cells from ALS patients, such as those of the motor neuron type, can be used to identify drug candidates for ALS treatment. The present review focuses on existing medication and medical needs of ALS patients, advantages of using patients’ iPSCs, scope for improvement in the approaches of motor neuron differentiation and functional assays, and strategies for the evaluation of drug efficacy in ALS disease.
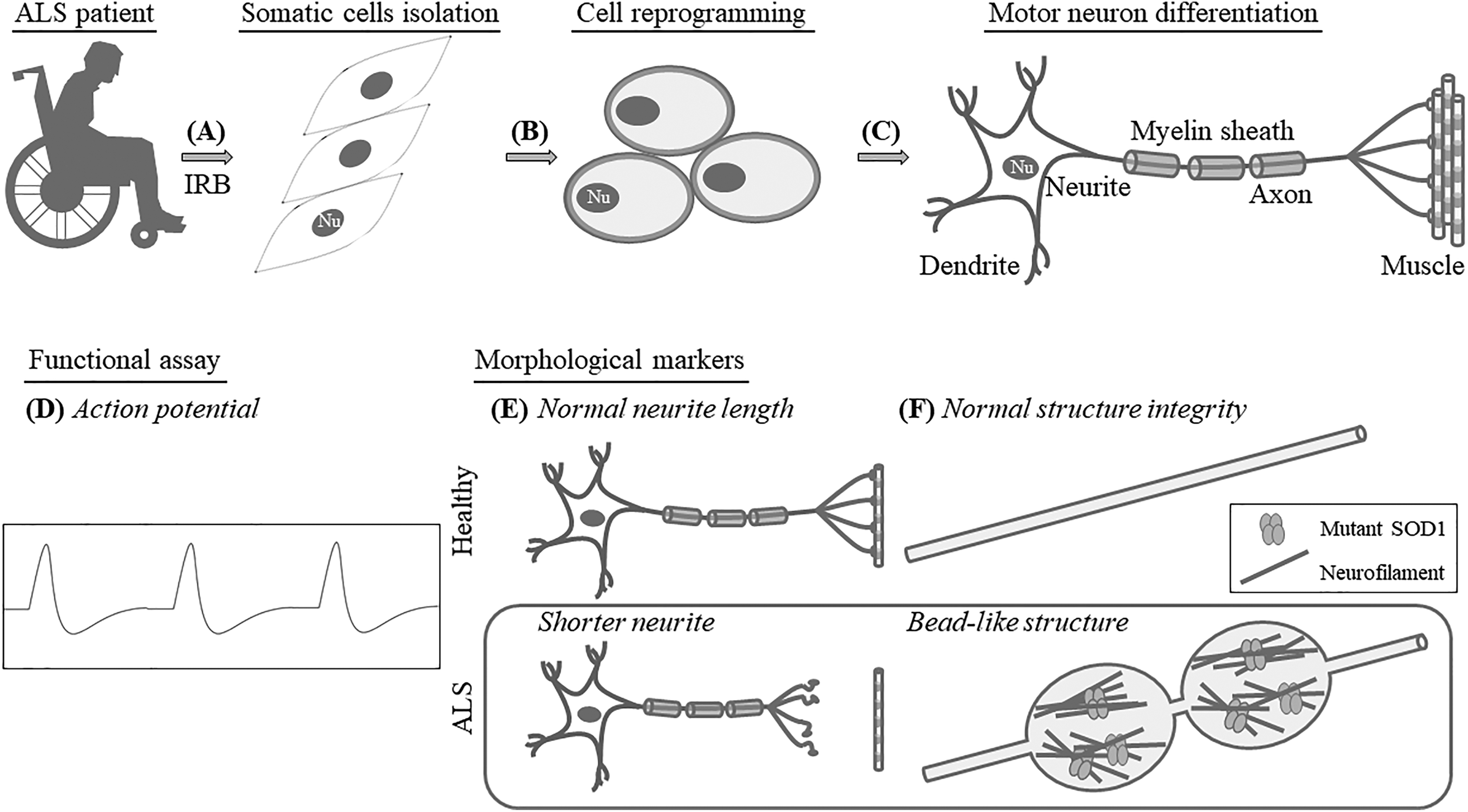
Schematic illustration showing that iPSCs generated from ALS patients differentiate into motor neurons for functional and morphological analysis. (A) ALS patients’ somatic cells are collected with donors’ informed consent under institutional review board monitoring (A) and the cells can be reprogrammed to the induced pluripotent state (B). After motor neuron differentiation of ALS patient’s iPSCs (C), further studies of the disease can be performed to assess physiological properties (D), providing a link to the level of maturation. The examination of morphological changes such as neurite length (E) subcellular aggregate (F), and patient-specific samples can be used to characterize the pathogenesis of ALS in patient-derived iPSCs. Nu: nucleus.
Currently Available Treatments and Potential Small Molecules
Drugs Approved by the U.S. Food and Drug Administration for ALS Treatment
Riluzole is the first available compound known to attenuate the progression and partly relieve the symptoms of ALS via depressing N-methyl-D-aspartate (NMDA) receptor- and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor-mediated signaling 43,44 . Riluzole has several side effects and extends survival only by an average of 2–3 months, so more effective ALS therapy is urgently needed 45,46 . The recently approved molecule edaravone is a small-radical scavenger that can inhibit oxidative stress. Edaravone was initially used to treat acute ischemic stroke 47 . It quenches the hydroxyl radical functional group and downregulates both radical-dependent and radical-independent lipid peroxidation 48 . Previous results showed that edaravone could delay the degeneration of motor neurons in a murine model with mutated SOD1 expression 49 and data from a phase 2 trial in ALS patients consistently reported the effect of the drug in reducing oxidative stress 50 . Although evidence supported these positive outcomes in a phase 3 trial of edaravone, the inconvenience of intravenous administration and the extremely restricted population of ALS patients caused physicians not to prescribe the drug 51 . In addition to the inconvenience and high treatment cost, the long-term efficacy and toxicity profile of edaravone have not yet been established. Therefore, the clinical needs and challenges in clinical trials for ALS patients still exist and much more efficient protocols for drug screening are required.
Small-Molecule Development for Clinical Application Targeting ALS
Because of extensive studies investigating small molecules, exploring mechanisms, and synthesizing new compounds, the scope of drug discovery is changing and scientists can combine the power of synthetic chemistry with advanced screening technologies for small-molecule development. These small molecules can be covalent bonding inhibitors, protein–protein interaction modulators, protein conformational allosteric regulators, or epigenetic controllers and they sometimes have effects on multiple targets at one time 52 –54 . For example, olaparib, an ADP-ribose polymerase inhibitor, and imatinib, a tyrosine kinase inhibitor, can significantly reduce ovarian cancer and chronic myeloid leukemia cells, respectively 55,56 . Furthermore, small molecules can potentially penetrate the blood–brain barrier into the central nervous system to treat patients with brain tumors or neurological diseases 57 . Creating small molecules that target mutant proteins associated with ALS may be promising candidates for the treatment of ALS.
Drug development based on neuroprotective properties such as reducing oxidative stress and excitotoxicity and eliminating toxic aggregates/fragments may benefit ALS patients. Recently, several groups have shown that autophagy plays an important role in ALS pathogenesis 58 –60 . The autophagy–lysosome machinery is the major system for removing abnormal aggregates through cytoplasmic protein degradation 61 . One of these molecules that has been preferentially examined using in vitro and in vivo ALS models and shows promise is a purified compound from a plant used in traditional Chinese medicine (Angelica sinensis), n-butylidenephthalide (n-BP). Evidence has suggested that accumulated mutant SOD1 can initiate autophagy in an animal model 59 and n-BP can extend the lifespans of the SOD1 murine model through modulating cellular autophagy and apoptosis in the spinal cord 60 . Furthermore, findings also indicated that n-BP could reduce the death of motor neurons, relieve muscular atrophy, suppress neuroinflammation, and recover gastrocnemius muscle 59 . Although the functions of n-BP in anti-apoptosis, anti-inflammation, anti-oxidation, and autophagy modulation have been revealed, the data were collected from mutated SOD1 in in vitro and in vivo models. Investigations based on ALS patients’ iPSCs may provide further information for the development of this drug candidate.
iPSCs Differentiate into Functional Motor Neuron Units
Differentiation into Motor Neurons
iPSCs can generate different subtypes of neural cells, such as neurons, motor neurons, astrocytes, and other cell types 35,62 –64 ; therefore, the characterization of these in vitro cells recapitulating in vivo ALS progression can be achieved. Previous studies reported that human ESCs have been differentiated successfully into motor neurons by exogenously expressing the mutated genes of ALS. The approaches of motor neuron differentiation have also been applied to iPSCs. fALS/sALS-derived motor neurons can be harvested and investigated for the development of new drugs (Fig. 1C). Crucial secreted signaling factors, such as bone morphogenetic protein (BMP), fibroblast growth factor (FGF), retinoic acid (RA), Sonic hedgehog (Shh), and Wnts and transforming growth factor-beta (TGFβ) signaling, which are involved in the patterning neural axis during brain development, have been used to direct iPSCs toward motor neurons 65 –68 .
Motor neurons are classified as corticospinal upper motor neurons and spinal cord lower motor neurons (LMNs). The degeneration and death of motor neurons lead to the onset of neurodegenerative diseases such as ALS 69 –71 . According to results obtained in different species 67,72 –75 , signaling in the development of neuroectoderm has been shown to specify LMN fate 66,68 . The process of motor neuron differentiation is time consuming, expensive, and inefficient in the acquisition of cells. After making efforts to fine-tune the protocols, two small molecules, LDN193189 and SB431542, were found to accelerate the differentiation of motor neurons through the inhibition of both BMP and TGFβ signaling and blocking mesoderm and endoderm fates 76 . Therefore, more abundant motor neurons can be obtained from iPSCs in a shorter time period.
Functional Assay and Morphological Marker for Motor Neurons
The most extensive functional examination of differentiated motor neurons is detection of the electrophysiological action potential for evaluating the maturation of motor neurons in vitro 76 (Fig. 1D). During the early stages of neural differentiation, voltage-gated ion channels need to be induced to generate an action potential. In addition, spontaneous action potentials require time to develop after the maturation of motor neurons 77 . To achieve a high level of mature motor neurons, the transition between induced and spontaneously occurred action potentials requires the involvement of various factors such as synapse formation 78,79 . To determine the propagation of electrical signals, protocols to direct the differentiation of motor neurons from iPSCs with the capability of synapse formation is critical.
Because ALS pathogenesis presents heterogeneous causes accompanied by distinct mechanisms linked to the onset and progression of ALS, imaging-based approaches are required to determine cellular/molecular changes and drug effects on ALS disease 80 . For evaluation, information on cellular morphology can be obtained by automated data acquisition, thereby lowering the cost of drug screening in terms of labor. Both the fragmentation of neurites with reduced length and morphological swelling, the so-called bead-like structure resulting from abnormal protein aggregation (Fig. 1E and F) 22,81 –83 , can be analyzed under the microscope. Such findings suggest that specific antibodies can be also used to label mutants for understanding the disease progression of ALS 84 . These protein aggregates are expressed in vivo in native form, so the antibodies may not be available to recognize targets correctly and represent the real distribution or localization.
Use of ALS Patient-Derived iPSCs for Drug Development
Drug-Screening Platform via iPSCs from ALS Patients
During the process of novel drug development, safety and efficacy testing are costly and time consuming. Many clinical trials of ALS have been withdrawn or failed due to unexpected toxicity or lack of efficacy 85 . The potential toxicities challenging clinical translation of drug candidates suggest that in vitro and in vivo nonhuman models might not be sufficient. To select effective therapies for ALS patients and to improve the success rate of drug selection, iPSCs have been used as models in many outstanding studies (Fig. 2A and Table 1) 2,23,34,35,52,58,83,86 –90 . Egawa et al. recently reported an iPSC drug-screening assay in which they used several compounds targeting the epigenetic/transcriptional level of iPSC-derived motor neurons from ALS patients with the TDP-43 mutation 2 . One of the compounds, anacardic acid, was shown to reverse part of the ALS phenotype found in motor neurons. This work can be a stepping stone to toward using ALS-specific motor neurons to verify candidate drugs.

Combination of human iPSC approach, high-throughput drug screening, and biomarker application can potentiate clinical candidate selection. (A) iPSCs derived from patients with ALS can be used for high-content drug screening to identify pathways and targets of small molecules with therapeutic potential. After preclinical studies, these small molecules may enter clinical trials with suitable administration routes (B) and the use of biomarkers can be used to calculate and determine drug efficacy (C). P-NFH: phosphorylated heavy chain of NF.
ALS Cell Models from Disease-Derived iPSCs.

WB: Western blotting analysis; IF: immunofluorescence assay; FISH: fluorescence in situ hybridization; IHC: immunohistochemistry; FRAP: fluorescence recovery after photobleaching; Isl-1: Islet-1; HB9: Homeobox 9; Tuj1: beta III tubulin; SMI32: neurofilament H non-phosphorylated; CHAT: choline acetyltransferase; VGLUT1: vesicular glutamate transporter 1; NMDAR1: NMDA receptor 1; SYT1: synaptotagmin 1, SYP: synaptophysin; ALDH1: aldehyde dehydrogenase 1; GFAP: glial fibrillary acidic protein; ASO: anti-sense oligo-nucleotide; CDK: cyclin-dependent kinase; JNK: c-Jun N-terminal kinase
Motor neurons derived from patients with fALS or sALS were used to screen various kinase inhibitors, epigenetic modulators, anti-sense oligo-nucleotides (ASOs), or approved molecules for any other indication (Table 1). Furthermore, Wainger et al. used a high-throughput electrode array to detect the action potentials of variant ALS patients’ motor neurons (SOD1, C9ORF72, and FUS mutation) and revealed that retigabine could reduce the death of motor neurons by blocking hyperexcitability 89 . The drug candidates identified by such methods can confirm the toxicity and efficacy of drugs in humans and retigabine has entered a phase 2 trial for efficacy evaluation (NCT02450552; estimated completion date: February 2018). Importantly, ALS-patient-specific iPSCs present many benefits, including the correct genetic background and pluripotency. The results from the above-mentioned drug-screening studies have shown that high-throughput approaches can produce abundant and useful information that can expedite and increase the probability of success for drug candidates entering clinical trials 91 (Fig. 2B).
Although iPSCs are mainly generated from skin fibroblasts or peripheral blood from ALS patients, the pathogenic phenotypes may disappear. These iPSCs may be reverted to the early stage pluripotent cells. Thus, the recapitulation of a late onset of neurodegenerative disease cannot be fully achieved 92 . To generate iPSCs associated with the characterization of aging, oxidative stress, DNA-damaging and proteasome modulator agents are being tested to produce high-level maturation of patient-specific iPSC derivatives 93 –95 . Previous reports indicated that AMPA receptor, a glutamate receptor, could mediate excitatory transmission in the brain; therefore, glutamate insult accelerates the aging process by allowing high level of calcium ions to enter the neurons 15,96 . Such induction can be used to change the phenotypes of patient-derived iPSCs to mimic neurodegeneration. In addition, downregulation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) can attenuate neuron death by manipulating the trafficking of the GluR2 subunit of the AMPA receptor within the cell membrane 97 . Many small molecules modulating PTEN have been synthesized, so it may be a potential therapeutic strategy for medical intervention in ALS patients.
To verify the use of screening approaches and to develop reliable platforms for exploring the molecular underpinnings of ALS, multiple iPSC clones from different ALS patients and several markers are urgently required. Using at least two iPSC lines from patients with ALS in combination with automated and imaging-based techniques would acquire considerable information on cell morphology, formation of protein aggregates, and distribution/patterns of specific molecules by immunocytochemistry/immunocytochemistry or immunofluorescence staining. Moreover, high-throughput equipment has been used for examining small-molecule compounds in several studies. Nevertheless, there is no clear mechanism of neuron death that can clearly distinguish sALS from fALS. Large sample sizes are necessary to reveal the detailed and unique ALS pathogenesis, but the cost of reprogramming ALS patients’ fibroblasts for iPSCs remains high.
Biomarkers of Motor Neurons
Throughout the drug development process, biologically validated biomarkers are used to assess the subject’s response to a specific treatment. Biomarker analysis demonstrating the effect of the drug candidates on ALS targets is also important (Fig. 2C). Previous studies showed that CD40L, which is expressed by activated T cells and required for induced immune response, could be a treatment target for ALS patients. Moreover, inhibiting CD40L-mediated inflammatory responses can improve survival in animal studies 98,99 . In addition to altered inflammatory gene expression, monocyte-expressed, ALS-specific, inflammation-related microRNA or the unregulated complement system can be treated with specific antibodies or inhibitors to delay ALS progression, extend survival, and improve motor function in animal models 100,101 .
Abundant neurofilament (NF) proteins have been detected in the cerebral spinal fluid (CSF) or blood from SOD1 G93A mice 102 –104 (Fig. 2C). Among them, plasma containing the phosphorylated heavy chain of NF was thought to be associated with motor neuron death 105,106 (Fig. 2C). Moreover, NF aggregates (bead-like structure) can also be observed in ALS-patient-specific iPSCs with mutation of SOD1 A4V or SOD1 D90A 81 (Fig. 1F).
Several studies have revealed that translational products of C9ORF72 repeats resulted in aggregation and were localized in the neurons of the central nervous system. This phenotype was seen in iPSC models 107,108 . When small molecules bind to the GGGGCC repeat sequence, they can inhibit the formation of translational aggregates in the differentiated motor neurons derived from iPSCs (Table 1). In addition, peptide aggregation has been found in CSF from ALS patients with the C9ORF72 mutation 108 (Fig. 2C); therefore, this measurement is available for evaluating therapeutic efficacy.
Other reports about genetic mutations also found the formation of TDP-43 inclusion bodies, which was used as one of the biomarkers 2,34,109 . Using enzyme-linked immunosorbent assay-based approaches, the TDP-43 level was found to be increased significantly in plasma and CSF from patients with ALS compared with healthy individuals. Although the discovery of biomarkers to distinguish ALS patients and healthy people are highly challenging, much more effective therapies may be investigated and developed with more biomarkers
Conclusion
Neurodegenerative diseases have a potentially disastrous impact on quality of life, so ongoing efforts to develop novel therapeutic drug candidates are of utmost importance 99,110 –113 . The use of genetic manipulation in cell systems and nonhuman animal models may not predict the potential toxicity and lack of efficacy accurately in humans 85 . Recent achievements in differentiation of patient-specific iPSCs into motor neurons have provided the basis to establish a platform to screen drug candidates, test toxicity, and confirm the efficacy of drugs in complicated fALS/sALS populations (Table 1). Although some constraints, such as the labor-intensive and high-cost procedure, protocols for generating fully mature motor neurons, detection methods of protein aggregates, applicable biomarkers, and basic mechanism of motor neuron death in sALS, need to be improved, the use of iPSC-dependent screening may accelerate the process of drug development. Following a systemic assessment (Fig. 2), studies of ALS may shed light on causes of disease and help to improve drug development in the future.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Everfront Biotech Inc.