
Abstract
Many neurodegenerative diseases are progressive, complex diseases without clear mechanisms or effective treatments. To study the mechanisms underlying these diseases and to develop treatment strategies, a reliable in vitro modeling system is critical. Induced pluripotent stem cells (iPSCs) have the ability to self-renew and possess the differentiation potential to become any kind of adult cell; thus, they may serve as a powerful material for disease modeling. Indeed, patient cell-derived iPSCs can differentiate into specific cell lineages that display the appropriate disease phenotypes and vulnerabilities. In this review, we highlight neuronal differentiation methods and the current development of iPSC-based neurodegenerative disease modeling tools for mechanism study and drug screening, with a discussion of the challenges and future inspiration for application.
Keywords
Induced-Pluripotent-Stem-Cell-Based Therapies and Neurodegenerative Disease Modeling
Aging societies have a number of health issues, particularly regarding neurodegenerative diseases such as dementia, Alzheimer’s disease (AD), and Parkinson’s diseases (PD). During the progression of these diseases, patients may lose their memory and thinking abilities, and many can develop movement disorders. However, the majority of neurodegenerative diseases lack effective treatments. For novel drug screening, various cell lines and animals have been used as neurodegenerative disease models. After revealing specific disease mechanisms, drugs designed to target pathogenic candidates can be developed. However, differences between disease models and the actual human nervous system continue to be a risk of disease modeling.
The first mouse iPSCs were established in 2006 in Dr Shinya Yamanaka’s laboratory 1 . Somatic cells were reprogrammed into early embryonic-like pluripotent stem cells (PSCs) via the re-expression of four transcription factors, Oct4, Sox2, Klf4, and c-Myc. These cells displayed a self-renewal ability and pluripotent differentiation potential (i.e. the ability to differentiate into ectoderm, mesoderm, or endoderm) in vitro. In 2007, the same research group successfully established a human iPSC line from skin fibroblasts 2 . Subsequently, many research groups have developed methods to establish iPSCs from numerous somatic cell sources including fibroblasts, adipocyte stem cells, neural stem cells (NSCs), hematopoietic stem cells (HSCs), and peripheral blood mononuclear cells 3 –6 . The advantages of these approaches have expanded the application potential of pluripotent stem cells without the source limitation or ethical concerns. In the next decade, iPSC technology has attracted increasing attention worldwide, and researchers have established and banked numerous iPSC lines for developmental study, disease modeling, genetic/epigenetic studies, and transplantation therapies (Fig. 1).

Applications of induced pluripotent stem cells.
For cell transplantation therapies, iPSC and embryonic stem cell (ESC)-derived lineage-specific cells have been applied to age-related macular degeneration (AMD), PD, heart disease, spinal cord injury, blood transfusion, cancer, and arthritic disorders. Clinical trials with PSC-derived (including iPSC and ESC) cells for AMD, PD, spinal cord injury, diabetes, and myocardial infarction are under progress 7 .
For neurodegenerative disease modeling, the greatest challenge is arguably the difficulty in obtaining disease-related tissue and cells directly from patients for pathology and physiology studies. For in vitro and in vivo modeling of neurodegenerative diseases, several cell and animal models have been developed. However, the majority of neurodegenerative disease models are based on artificial cells or animals. For example, pathogenic-gene-overexpressed models are widely used for AD, PD, amyotrophic lateral sclerosis (ALS), and spinocerebellar ataxia (SCA) studies. However, these overexpression models show different cytopathology and disease mechanisms when compared with patient brain neurons, and the differences between animal and human brain remain one of the biggest challenges of animal-based brain disease models. Furthermore, animal models of neurodegenerative diseases may take a long time to recapitulate phenotypes and are also time and resource consuming for drug screening. The iPSC modeling system allows studies to use patient cell-derived pathogenic cells to address disease phenotypes and their progression in a cell culture dish. Compared with other models, patient cell-derived iPSCs may serve as a reliable in vitro disease model of complex neuronal diseases. This model may serve as an accurate first line for drug screening and candidate exploring before animal models. Many reports have successfully established iPSC lines from patient tissues for various neurodegenerative diseases such as AD, PD, ALS, SCA, Rett syndrome, spinal muscular atrophy (SMA), Down syndrome (DS), and Huntington’s disease (HD). In some cases, patient iPSC-derived neurons recapitulate disease phenotypes, such as amyloid-β (Aβ) aggregates and neuronal function degeneration that are seen in AD and can be applied to drug screening and mechanism discovery 8 –46 .
Induced-Pluripotent-Stem-Cell Establishment, Culturing, and Neuronal Differentiation
Induced-Pluripotent-Stem-Cell Establishment and Culturing
The technology for establishing iPSCs is improving every day. In the beginning, retrovirus and lentivirus vectors were used for the delivery of reprogramming factors. However, the integrative property of retroviruses may be a concern for genetic stability. For an integration-free delivery system, piggyBac transposons 47 , RNA viruses 48 , episomal vectors 49 , RNAs 50 , and proteins 51 have been used to replace integrative viruses. To improve iPSC generation efficiency, small molecules with signaling activities, as well as DNA demethylation and deacetylation, can robustly enhance iPSC colony-formation rate 52 –54 . Dr Hou’s research group developed a reprogramming method with only chemical compounds 55 . Recently, epigenetic modulation methods have been developed to generate iPSCs 56 .
The traditional PSC culture, including those of ESCs and iPSCs, consists of a coculture with fibroblast feeder cells 57 . For cell viability, avoiding single-cell dissociation is a common approach when passaging PSCs 57 . However, the feeder cell coculture system can become a challenge for cell property analysis, and dissociated cell death restricts cell clonal purification. Recently, many feeder-free and xeno-free culture systems have been reported to support the long-term growth of PSCs. Commercialized medium including mTeSR, Essential 8, PSGro, L7, and StemFit have been combined with coating matrix Matrigel, Geltrex, vitronectin, synthemax, laminin 521, and laminin E8 58 –61 . These culture systems have eliminated the contamination of feeder cells and animal serum. In addition, it has been discovered that the Rho/ROCK signaling pathway plays major role in dissociation-induced cell blebbing in PSCs 62,63 . This finding provides the possibility for single-cell dissociation and has expanded the PSC application aspects to genome editing, clonal isolation, and single-cell characterization.
Neural Differentiation
For neurodegenerative disease modeling, the differentiation of PSCs into candidate neural lineages is the key factor to recapitulating disease phenotypes. The differentiation protocol from PSCs to NSCs is dependent on human embryonic development (Fig. 2). Neuronal cells primarily come from a neuroectoderm lineage. To specifically differentiate PSCs into NSCs, the dual inhibition of the SMAD signaling pathway via the bone morphogenetic protein (BMP) and transforming growth factor beta 1 (TGF-β1) antagonists lead to robust neuroepithelial generation via inhibition of mesoendoderm formation 64 . The dual SMAD inhibition protocol converts PSCs into high purity forebrain NSCs with expression of Pax6, FOXG1, and Otx2 to form the cerebral cortex. For naïve cell fate, most NSCs induced via the dual SMAD inhibition method convert into forebrain cortical neurons. For other neural-type patterns, patterning factors are needed. In a previous study, researchers demonstrated that the combination of basic fibroblast growth factor (bFGF or FGF2), a TGF-β1 inhibitor, and a Wnt agonist promotes high efficiency NSC differentiation 65 .

Protocols of neural differentiation from pluripotent stem cells follow the mammalian central nerve system developmental process.
Specific Neural Lineage Patterning
During early embryonic neurogenesis, neuroepithelial cells form the neural plate and neural tube for the central nervous system (CNS). NSCs in the neural tube are patterned by ‘morphogens,’ which are dose-dependent developmental signaling factors, to generate and form the whole CNS 66 . In addition, sonic hedgehog (Shh) signaling plays a decisive role in dorsal-ventral patterning. To pattern neuroepithelial cells into ventral lineages, Shh, purmorphamine, or a Smo agonist needs to be added into the patterning medium, and treatment with the Shh antagonist cyclopamine can pattern neuroepithelial cells into a dorsal lineage. For rostral-caudal patterning, Wnt signaling, BMP signaling, and retinoic acid (RA) signaling, all participate in the neural fate decision. Thus, for in vitro CNS neuronal differentiation, morphogens or their agonists/antagonists are applied for specific neuron patterning. For midbrain dopaminergic neuron (DA neuron) differentiation, Shh and fibroblast growth factor 8b (FGF8b) are applied to pattern NSCs to ventral midbrain–hindbrain boundary (MHB) neurons, which is the primary localization of DA neurons 54,67 . FGF8b is highly expressed in MHB neurons during embryonic neural tube development 68 . However, DA neuron patterning efficiency in the FGF8b/Shh method is not good enough at only 10–30% overall. To improve DA neuronal patterning efficiency, the GSK3β inhibitor CHIR99021 is applied, which has been shown to greatly improve DA neuron patterning efficiency 69,70 . CHIR99021 is the first small molecule shown to dose-dependently inhibit GSK3β activation, a Wnt signaling downstream protein 71 . This small molecule has benefited many PSC in vitro differentiation studies that required different Wnt activation levels. Combining low dosage CHIR99021 with the patterning factors Shh and FGF8b enhances DA progenitor-specific markers, including FOXA2, Lmx1a, Nurr1, and Pitx3. The addition of CHIR99021 can also improve DA neuron generation efficiency up by more than 80% with the DA-specific markers TH, DAT, and dopamine secretion 70 . In the previous DA neuron differentiation two-step methods, PSCs are induced to NSCs and then they start the patterning process. A novel DA neuron differentiation process that induces neural differentiation and DA neuron patterning simultaneously has been shown to enhance DA neuron generation efficiency 69,70 . For DA neuron purity, cell surface specific markers are another key factor for isolating DA progenitors. The ventral neural tube-specific surface protein Corin 72 and the midbrain-specific surface marker LRTM1 73 can also be applied for fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS) for high purity DA progenitor isolation.
Motor neuron (MN) differentiation is a three-step protocol, with dual SMAD inhibition to convert PSCs to NSCs, activate RA signaling for caudalization, and activate Shh signaling for ventralization. For better MN generation efficiency, various protocols with different small compounds, treatment combinations, and time periods have been applied for MN patterning 74 . One breakthrough in MN patterning was the evaluation of the GSK3β inhibitor CHIR99021 on spinal cord neural lineage patterning. Maury et al. 75 and Du et al. 76 discovered that the addition of CHIR99021 to the traditional MN induction protocol greatly enhances the spinal cord specific markers CDX1, CDX2, HOXA4, and HOXA5. MN-specific marker expression enhances by 90% depending on the CHIR99021 dosage, including Oligo2, Islet1, and HB9 76 . Subsequently, various studies have demonstrated that GSK3β inhibition plays a key role in spinal cord MN differentiation; thus CHIR99021-based methods have become widely used.
For other specific neural lineage patterning, protocols have been established for gamma amino-butyric acid (GABA)-ergic interneurons 77,78 , serotonin neurons 79 , and glutamate neurons 80 , by following the basic neural tube development protocol and Wnt signaling level.
A very special neuron lineage with a high patterning challenge is cerebellum Purkinje cells, which represent the major relevant neurons related to SCA pathology. Purkinje cells are located at the cerebellar plate of dorsal MHB neurons. However, there is no efficient protocol to generate Purkinje cells from PSCs according to the neural tube development principle. Muguruma et al. 81 discovered that endogenous MHB patterning factors are more important compared with exogenous factors such as cyclopamine (a small compound Shh antagonist) or FGF8b. In mouse PSCs, the collective effect of bFGF and insulin can dramatically enhance MHB marker expression, including that of Wnt1, FGF8, EN2, and Gbx2. Furthermore, bFGF/insulin can also increase the early Purkinje cell markers Corl2, Neph3, and Ptf1a. After neuron maturation, Purkinje cells express the specific marker L7 and have classical Purkinje cell morphology and neuroelectronic response 81 . This differentiation method can also be applied to human PSCs. After bFGF/insulin treatment, about 20% of PSCs can become Purkinje cell progenitors. After Purkinje progenitor cell purification using specific surface markers and granule cell coculture (or brain slice coculture), mature Purkinje cells display classical neuronal function 24,82 .
Methods have also been developed for another important CNS cell lineage, glial cell differentiation. Two major kinds of CNS glia are astrocytes and oligodendrocytes. Astrocytes are multi-function cells that benefit neuron survival and function, including nutrition exchange, metabolism control, and immune regulation. Oligodendrocytes can myelinate neurites to protect CNS neurons. For astrocyte differentiation, PSC-derived NSCs are primarily induced with BMP, ciliary neurotrophic factor (CNTF), bFGF, and fetal bovine serum or serum-free medium with Activin A, heregulin 1β, and insulin-like growth factor 1 (IGF-1) 83 . For astrocyte purification, repeating trypsinizing passages serve to eliminate the majority of other neuronal cells 84 . Afterward, the purified astrocytes can be applied to disease modeling, neuron coculture, or neuron–astrocyte interaction studies.
Another key glial cell type is oligodendrocytes, and their differentiation is much more complex and challenging. In this process, there are six steps that take more than 180 days to obtain functional oligodendrocytes from PSCs 85 . The first step is to transfer PSCs into NSCs. Afterward, the Shh agonists, RA and bFGF help NPCs to become oligo2+/Nkx2.2+ oligodendrocyte progenitor cells (OPCs). For myelinogenic OPC differentiation, combined growth factor treatment for 120 days is needed. Finally, OPCs are transplanted into animals for terminal maturation. The resulting O4+ OPC purity is typically 4.1–11.9%.
Microglia are the macrophages of the CNS, where they play an important role in the CNS immune response. During early embryonic development, there are two origins of microglia, the early yolk sac and mesoderm-derived HSCs. Muffat et al. 86 developed a novel chemically defined method to differentiate PSCs into microglia via the yolk sac route. However, this protocol is complex for routine use. To address this issue, Pandya et al. 87 provided a concise two-step protocol to generate microglia. The first step consists of converting PSCs into HSCs with CD34 and CD43 expression. The second step involves the coculture of PSC-derived HSCs with astrocytes. After coculture, CD39+ microglia are sorted for use in subsequent experiments.
Overall, patterning methods that induce NSCs to differentiate into various types of specific neural lineages have greatly benefited PSC-based neurodegenerative disease modeling systems. However, some special kinds of neurons and glial cells present with unique technical difficulties for routine generation and application to disease modeling.
Current Neurodegenerative Disease Modeling and Drug Screening Using Induced-Pluripotent-Stem-Cell Models
The most serious neurodegenerative diseases, including frontotemporal dementia (FTD), AD, and PD, represent the major targets of iPSC-based disease modeling. iPSC-based in vitro models have also been applied to some rare diseases such as DS, ALS, SMA, HD, and SCA. The major publications resulting from these studies are listed in Table 1.
Lists of typical publications of induced pluripotent stem cell-based neurodegenerative disease modeling.

α1ACT: C-terminal of CaV2.1; AChR: acetylcholine receptor; AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; Aβ: amyloid beta; APP: amyloid precursor protein; BDNF: brain-derived neurotrophic factor; Bdph: N-butylidenephthalide; CaV2.1: gene product of CACNA1A; DA: dopaminergic; DHA: docosahexaenoic acid; DS: Down syndrome; ER: endoplasmic reticulum; FB: forebrain; FUS: fused in sarcoma gene; GSK3β: glycogen synthase kinase 3 beta; HD: Huntington’s disease; HDAC6: histone deacetylase 6; Htt: Huntingtin; MN: motor neuron; NSCs: neural stem cells; PD: Parkinson’s disease; PMOs: phosphorodiamidate morpholino oligonucleotides; p-Tau: phosphorylated Tau protein; ROS: reactive oxygen species; SCA: spinocerebellar ataxia; SMA: spinal muscular atrophy; SMN: survival motor neuron; SP: sporadic; TGF-β: transforming growth factor beta; TH: thyroid hormone; TRH: thyrotropin-releasing hormone; UBA1: ubiquitin-like modifier activating enzyme 1; UPR: unfolded protein response; VAPB: vesicle-associated membrane protein-associated protein B; VPA: valproic acid.
Induced-Pluripotent-Stem-Cell-Based Modeling for Alzheimer’s Disease
Amyloid accumulation and Tau protein abnormalities are the two major cytopathies observed in AD patient brain neurons. To recapitulate AD cytopathies in an in vitro modeling system, DS and AD iPSC-derived cortical neurons have been applied to mimic AD for mechanism studies and drug screening 11,12,25,29,30,34,37,41 –44 . DS neurons express some classical AD cytopathies, including Aβ42 aggregations, Aβ40 over-secretion, Tau protein overexpression, hyperphosphorylation, and redistribution 41 . Moreover, DS forebrain neurons have reduced synaptic activities and show vulnerability to oxidative stress 43 . Interestingly, studies have also found that DS astroglia cannot support neurogenesis 12 . Because of this strong phenotype expression, DS models are suitable for AD drug screening. In our previous study, we found that the herbal compound N-butylidenephthalide decreases Aβ and Tau protein cytopathies 11 . However, the genetic backgrounds of DS and AD are largely diverse, suggesting that the accuracy of DS-based AD modeling may be another challenge. Cortical neurons derived from familial and sporadic AD iPSCs have amyloid and Tau protein abnormalities, including Aβ accumulation, Aβ42/Aβ40 ratio dysregulation, and Tau protein hyperphosphorylation 25,29,30,34,37,42,44 . In some reports, increased reactive oxygen species and apoptosis signaling have been observed in AD neurons 29 . Interestingly, Kondo et al. 29 demonstrated that early AD phenotypes within different kinds of familial and sporadic AD neurons are varied. One candidate AD compound, docosahexaenoic acid (DHA), was shown to only rescue some types of AD neurons and showed no effect on others. This study demonstrated that separating AD sub-populations may be helpful for selecting the right drug for targeting the specific AD types for effective personal therapy. The same research group also established an AD platform for systemic drug screening. In this study, they found that a set of combined anti-Aβ cocktails may inhibit cytotoxic Aβ accumulation in both familial and sporadic AD neurons 30 .
Induced-Pluripotent-Stem-Cell-Based Modeling for Parkinson’s Disease
PD is another serious neurodegenerative disease that affects patients all around the world. The number of DA neurons decrease 5–10% per decade due to the natural aging process and are majorly induced by reactive oxygen species (ROS) stress and metabolism alterations 88 –90 . However, the DA neurons are largely dead in the PD patient’s brain within a few years and cause serious movement disorders. For PD in vitro modeling, familial (including LRRK2, SNCA, PINK1, and PARK2 mutation) and idiopathic PD iPSCs have been differentiated into DA neurons and applied to cytopathic studies 15,16,32,38,39 . In addition, α-synuclein accumulations have been observed in LRRK2 mutations and SNCA triplication DA neurons 16,38 . In PINK1 and LRRK2 mutant DA neurons, mitochondrial dysfunction and DNA damage were both observed 15,39 . For idiopathic PD modeling, DA neuron degeneration, Lewy-body accumulation, mitochondria deficiency, and autophagy dysregulation were also found in sporadic and progerin-induced aging DA neurons 32,38 . Taken together, both familial and idiopathic PD iPSC-derived DA neurons had late disease phenotypes such as neurite degeneration and cell apoptosis. However, the expression of specific early cytopathies such as α-synuclein accumulations were restricted to familial PD with appointed genetic mutations.
Induced-Pluripotent-Stem-Cell-Based Modeling for Amyotrophic Lateral Sclerosis
ALS is arguably the most concerning, rare neurodegenerative disease. For ALS modeling, patient iPSCs have been differentiated into MNs, astrocytes, and oligodendrocytes for modeling 8,10,13,18,19,21 –23,27,33,40,46 . Sporadic and TDP-43 mutant MNs show cytopathies, including cytosolic TDP-43 aggregations, neurite degeneration, and MN death 10,18,23 . In addition, SOD1 gene mutation MNs have been shown to express SOD1 protein aggregates and neurofilament dysregulation phenotypes 13 . Disrupted nucleocytoplasmic transport has been identified in c9orf72-mutant MNs 46 . In FUS-gene-mutant MNs, FUS protein mislocalization, cellular vulnerability, and axonal transport defects were also observed 21,22 . Imamura et al. 23 screened more than 1000 candidate drugs and identified that the Src/c-Abl pathway may be a potential therapeutic target to benefit MN survival in many kinds of familial and idiopathic ALS MNs. Besides MNs, researchers have also found that astrocytes and oligodendrocytes derived from TDP-43 and SOD1 mutant iPSCs cannot support MN maturation or survival but do contribute to MN death, just as DS astroglia on cortical neurons 19,40 .
Induced-Pluripotent-Stem-Cell-Based Modeling for Rare Neurodegenerative Diseases
Aside from the examples described above, iPSC-based modeling has also been applied to rare neurodegenerative diseases such as HD 9,14,26,35 , SCA 24,28,36 , and SMA 17,20,31,45 . The down-regulation of the proteasome, autophagosome, cadherin, TGF-β1, and brain-derived neurotrophy factor, as well as increases in Huntingtin protein aggregates, have all been identified in HD NSC and GABAergic neurons. These cytosolic abnormalities appear to contribute to apoptosis signaling activation, cytosolic stress vulnerabilities, and neural death of HD GABAergic neurons. In SMA MNs, decreased functional SMN protein leads to neurite degeneration, excitability, and neuromuscular junction dysfunction. For SCA modeling, neurons and Purkinje cells have been used to recapitulate SCA phenotypes. Ataxin 3 protein aggregates and autophagy decreases are both observed in type 3 SCA neurons 28,36 . Ishida et al. 24 successfully differentiated iPSCs into functional Purkinje cells and found specific cytopathies, including CaV2.1 (gene product of CACNA1A) up-expression, C-terminal of CaV2.1 (α1ACT) down-regulation, and vulnerability to triiodothyronine (T3) depletion in type 6 SCA. Compounds such as reluzole and thyrotropin-releasing hormone may reverse T3-depletion-induced neurite degeneration in Purkinje cells.
iPSC-based neuronal models have not only been used for neurodegenerative disease modeling but also epidemic studies. One study found that iPSC-derived neurons from twins may have different Zika virus affinity and infection rates in that one of the twins was infected with the Zika virus, but the other one was not. This study suggests that Zika virus infection may be correlated with the genotype or specific membrane receptors of individuals 91 .
Organoid and Coculture Systems Improve the Integrity of In Vitro Modeling
iPSC-derived neurons from patients may be the closest model for diseases. However, single-cell type in vitro models may be not enough to reproduce complex diseases such as neurodegenerative diseases. Indeed, specific interactions within many cell types such as neurons, glial cells, microglia, and connective tissue may underlie the progression of neurodegenerative diseases. Therefore, many coculture and organoid methods have been developed for whole-map analysis of neurodegenerative diseases.
Eiraku et al. 92 first demonstrated that three-dimensional (3D) cultures of neuronal embryoid bodies (EBs) can form primitive neural-tube-like structures in 2008. In 2011, the same research group differentiated human ESCs with the EB method to establish eye-cup-like organoids 93 . Later, Kadoshima et al. 94 discovered the forebrain-like development and structure in neural EBs. The neuroepithelial cells formed a lumen similar to the ventricular and the NSCs that expressed N-cadherin and CD133 were surrounded by the ventricular. This finding is similar to the NSCs in the sub-ventricular zone (SVZ) of the brain. Lancaster et al. 95 developed a simple 3D culture system to enlarge brain organoids in 2013. They embedded neural EBs in Matrigel droplets and used a spin culture system to force EB growth to become a well-organized brain organoid. These brain organoids could form multi-layer cortical-like structures after months of culture. Moreover, the neural division and migration in the organoid followed that of brain development, such as interkinetic nuclear migration. After applying brain organoids to disease research, authors mimicked inherited microcephaly and found reductions in NSC division in the SVZ-like zone of patient organoids. For further application, brain organoids have also been used for epidemic disease studies. iPSC-derived brain organoids were used to study Zika virus infection, and it was found that the Zika virus induced premature differentiation of neural progenitors, followed by microcephaly 96 . This novel system has allowed investigators to mimic neural development and migration in the human brain and to realize the actual situation of brain diseases. Subsequently, several publications have demonstrated organoids of cortical brain 97 , dorsomedial telencephalic tissue 98 , midbrain 99,100 , cerebellum 82 , and neural tube 101 . However, whether these organoids can be applied to developmental and disease modeling still needs evaluation.
For neurodegenerative diseases, the cytopathies may not lie only within neurons. Indeed, surrounding glial cells such as astrocytes, oligodendrocytes, and even immune cells may aggravate the progress of these diseases. Therefore, several publications have suggested that astrocytes derived from the iPSCs of patients with DS and ALS revealed deficiencies in their ability to support neuronal maintenance and even displayed toxicity to healthy neurons 12,40 . ALS patient iPSC-derived oligodendrocytes have also been reported to hurt MNs in vitro 19 . Although researchers have developed methods to directly differentiate PSCs into microglia, microglia differentiation remains a formidable challenge for disease modeling. For some neurodegenerative diseases, such as ALS and SMA, muscle cells that form neuromuscular junctions are important to recapitulate disease phenotypes in vitro 45 .
Combining Genome Editing Technology with Induced Pluripotent Stem Cells for Disease Modeling
Isogenic control is extremely important for accurate neurodegenerative disease modeling. The generation and correction of single-site DNA mutations benefit studies of genetic function and novel single nucleotide polymorphism (SNP) discovery. Most common genetic modification strategies include the use of enzymes to cut a nick at a specific DNA site 102,103 ; for DNA repair, cells would start non-homologous end joining or homologous recombination to generate DNA-point mutations or increase foreign DNA integration ratio for correction. To recognize specific sites on chromosomes, zinc finger nuclease (ZFN) 104 , transcription-activator-like effector nucleases (TALENs) 105 –107 , and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-1-associated system 9 (Cas9) 108 –110 have been applied for site-specific genome editing. Compared with DNA interactive protein (ZFN and TALEN)-based genome editing tools, RNA-based DNA pairing CRISPR/Cas9 is much easier to handle and has quickly become the most popular genome editing tool. Site-specific corrections have been widely used in AD 30 , PD 111 , ALS 13,23 , HD 9 , and other neurodegenerative diseases’ iPSC lines to have isogenic controls for research studies and large-scale drug screening. Genome editing technology is well established for generating site-specific knock-outs, mutant site modifications, polyglutamine (PolyQ) corrections, and large fragment deletions and insertions 112,113 in iPSC lines. For large DNA fragment modifications, a study was provided by Jiang et al. 114 . This research group introduced an inducible X-inactive specific transcript (XIST) gene into the DYRK1A locus of chromosome 21 and successfully silenced one copy of trisomy chromosome 21 on DS iPSCs to rebalance genetic expression level. Until now, CRISPR/Cas systems had only been applied to nicking DNAs or RNAs, but now they are being used for epigenetic regulations. After disrupting the DNA nuclease function, DNA methyltransferases (DNMTs) and methylcytosine dioxygenase have been fused to the catalytically inactive Cas9 (dCas) protein, which targeted the promoter regions to transiently turn on/off specific genes 115 . Combining these powerful tools, researchers are able to explore in more detail the mechanisms of neurodegenerative diseases.
Challenges of Induced-Pluripotent-Stem-Cell-Based Neurodegenerative Disease Models
Personal PSC-derived neurons have huge potential for modeling pathologies in a given patient’s brain. However, there are still many challenges that remain. Although iPSC technology was established over a decade ago, mimicking neurodegenerative diseases via iPSC-based models is still in infancy.
Until now, most models have been based on known genetic mutations. However, most high prevalence neurodegenerative diseases involve genetic mutations that are unknown or independent. For AD and ALS, less than 10% of patients have amyloid precursor protein (APP), secretase, ApoE, SOD1, TDP-43, or c9orf72 genetic mutations. Previous studies have also demonstrated that sporadic AD iPSC-derived cortical neurons do not show Tau-protein-related pathology. In some other cases, of neurodegenerative disease modeling with iPSCs, similar phenomena occur. These studies suggest the challenges to recapitulating disease phenotype with iPSC models. Therefore, large-scale input of familial and idiopathic disease iPSC lines for studies may provide improved data and experimental results to move the state of the science forward.
Aging is another serious issue for iPSC-based disease models. When reprogramming, most aging-related genes are turned off and the cells become original young cells. To accelerate disease phenotype expression, various treatments are added to the differentiated cells to increase oxidative stress, endoplasmic reticulum stress, mitochondria stress, ion channel stress, nutrition depletion, or autophagy inhibition. However, these relevant factors may be different with natural aging, thus decreasing the accuracy of iPSC-based disease models. Furthermore, how to discover the real relevance of pathogenic cells is still unclear. Progerin, a truncated form of lamin A protein that is involved in Hutchinson–Gilford progeria syndrome (HGPS), a premature aging disease, is found to accelerate cellular aging. Miller et al. 32 demonstrated that Progerin overexpression enhances aging and pathology of PD iPSC-derived midbrain DA neurons, including neurite degeneration and Lewy-body-like inclusions. This study may bring new insights for recapitulating the aging process of neurodegenerative diseases with iPSC models.
Another big challenge is the identification of the correlation between early abnormal phenomenon observed from iPSC-derived pathogenic neurons and real neuronal degeneration in the patient brain. Several reports have demonstrated an early pathologic-like phenomenon of iPSC-based neuronal disease models, such as Aβ42 and Tau abnormal in AD, TDP-43 distribution, neurofilament dysregulation, and nucleocytoplasmic transport disruption in ALS. However, it remains difficult to identify these early cytopathies as highly correlated with neural dysfunction, apoptosis, and cell death. Therefore, how to uncover detailed mechanisms that connect these early cytopathies with late-stage neuronal loss in culture and the patient brain may be the next step to overcome.
The neural differentiation process always takes at least a couple of months to obtain functional neurons for pathology studies, thus delaying the efficiency of iPSC-based drug screening. To accelerate neural differentiation and apply these findings to large-scale drug screening, inducible neurogenin 2, islet 1, and LIM Homeobox 3 have been introduced into iPSCs, which largely shorten the differentiation time of cortical neurons and MNs (less than 15 days to obtain functional neurons) 23,30 .
Furthermore, it still takes time to develop mature organoid and coculture systems to imitate complex nervous system tissue for detailed studies instead of neuron-only studies.
Future Aspects for Application of Induced Pluripotent Stem Cells in Neurodegenerative Disease Modeling
Besides stem cell techniques, big data, genome sequencing, and microarray technologies have been growing in leaps and bounds in recent years. By combining these novel technologies with iPSC disease models, unprecedented inspiration may also be freed (Fig. 3).
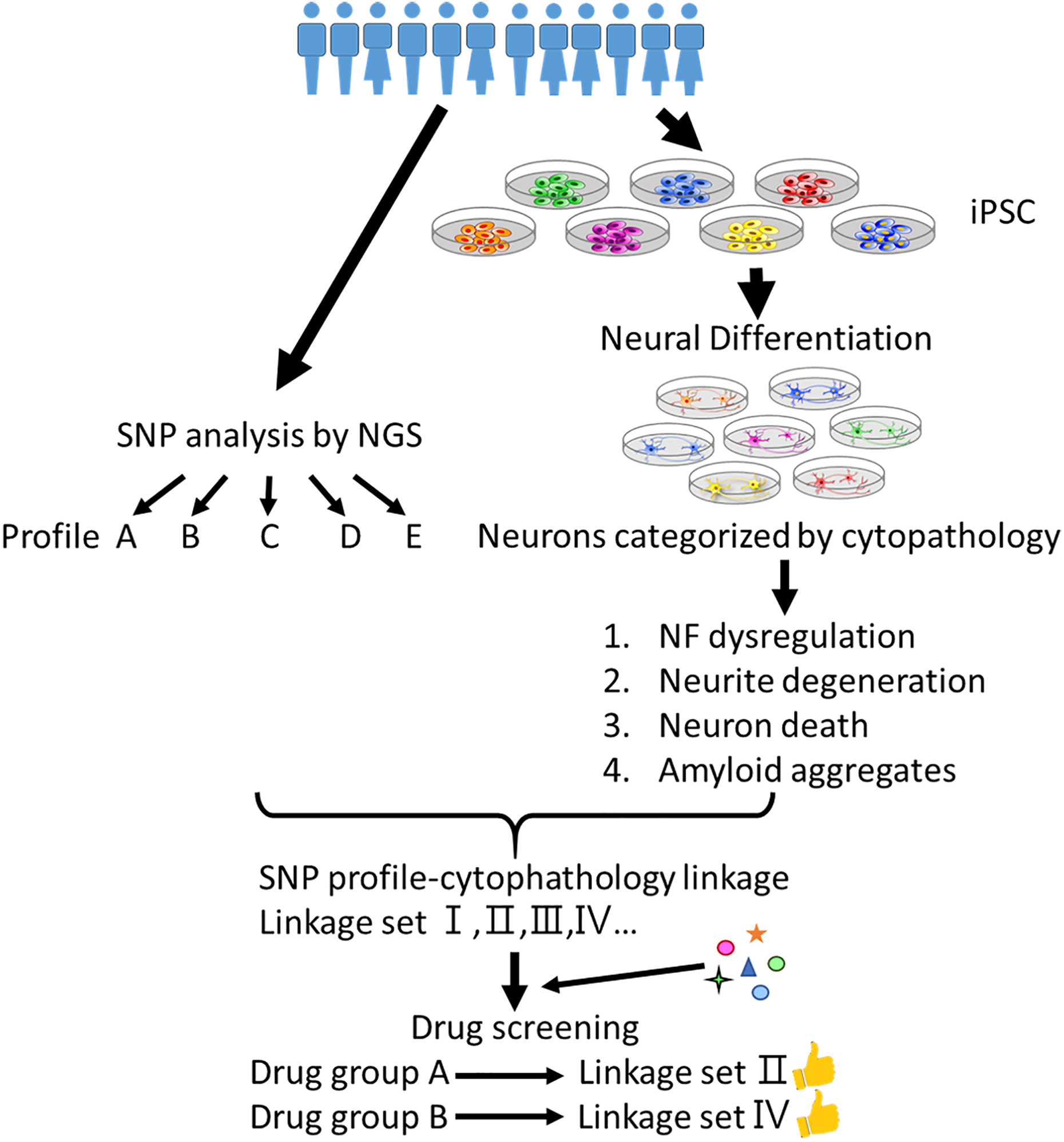
A combination of induced pluripotent stem cells, next-generation sequencing and big data technologies provides potential to develop precision medicine for neurodegenerative diseases.
Next-generation sequencing (NGS) provides efficient and low-cost full genome and transcriptome sequencing. Combining NGS with big data and iPSC models, researchers can share and compare sequencing and in vitro cytopathic data to reveal correlations between SNPs, transcriptomes, and disease phenotypes. This inspiration quickly brings genetics and cytopathies together to benefit disease studies.
Neurodegenerative diseases are so complex that detailed disease processes and mechanisms may be largely different between patients. Therefore, extensive drug screening with iPSC in vitro models may help us realize the relationship between candidate drugs and disease phenotypes, and indicate potential drug cocktails to treat specific groups of patients. Another potential solution is to establish personal iPSCs and select candidate compounds to these personal in vitro models before treatment. According to this idea, some drugs that have failed in clinical trials previously may still have potential if we can select suitable patients via in vitro pre-tests. After that, personal precise medicine may be applied for choosing the right drugs to fit the right patient.
In conclusion, although we still remain in the initial stages of understanding neurodegenerative diseases, the robust novel techniques described here may furnish us with the potential to better understand these complex and challenging conditions and develop appropriate treatment methods.
Footnotes
Authors’ Note
Chia-Yu Chang and Hsiao-Chien Ting are co-first authors.
Authors’ Contributions
SZL and HJH initiated this project. CYC edited, organized and wrote the article. HCT drew figures and tables. CAL and HCT wrote the text body. HLS and TWC organized and proofed the article. All authors reviewed this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Bio-innovation Center and financially supported by the Ministry of Science and Technology, Taiwan (MOST 104-2314-B-303-017-MY3, MOST 106-2320-B-303-001-MY3 and MOST 106-2320-B-303-002-MY3), Buddhist Tzu Chi Medical Foundation and Buddhist Tzu Chi General Hospital, Hualien, Taiwan.