
Abstract
Brain trauma is often associated with severe morbidity and is a major public health concern. Even when injury is mild and no obvious anatomic disruption is seen, many individuals suffer disabling neuropsychological impairments such as memory loss, mood dysfunction, substance abuse, and adjustment disorder. These changes may be related to subtle disruption of neural circuits as well as functional changes at the neurotransmitter level. In particular, there is considerable evidence that dopamine (DA) physiology in the nigrostriatal and mesocorticolimbic pathways might be impaired after traumatic brain injury (TBI). Alterations in DA levels can lead to oxidative stress and cellular dysfunction, and DA plays an important role in central nervous system inflammation. Therapeutic targeting of DA pathways may offer benefits for both neuronal survival and functional outcome after TBI. The purpose of this review is to discuss the role of DA pathology in acute TBI and the potential impact of therapies that target these systems for the treatment of TBI.
Keywords
Introduction
The sequelae of brain injury depend on the extent of injury as well as the system(s) that is directly or indirectly injured. Severe injury resulting from head trauma destroys anatomic structures in the brain and produces neuron and white matter loss. Severe injury is also associated with neurological deficits related to permanent neuronal circuit damage, so the therapeutic goals involve restoration of circuits, which is an area of active investigation. In contrast, mild to moderate injury may be related to indirect effects on neurotransmission that result from secondary brain injury. From a neurological surgery point of view, brain injury may result not only from trauma but also from the effects of surgical treatment, so investigations into mechanisms of brain injury may also help aid neurosurgical operations either for posttrauma or postsurgery care.
Many of the neurological deficits that result from brain injury are the result of direct anatomical damage, but suppression of functional electrical and chemical transmission also plays an important role. These changes have been investigated in epigenetic and behavioral studies.
1,2
Among neurotransmitter systems, dopamine (DA) pathways seem especially vulnerable to brain injury due to anatomic properties of the system, and there is considerable evidence that DA dysfunction contributes to posttraumatic brain injury deficits. First, cognitive dysfunction after traumatic brain injury (TBI) is associated with damage in the hippocampus,
3,4
striatum,
5
and frontal cortex,
6,7
which together mediate attention, executive function, learning, and memory
8
–10
; DA impacts each of these brain regions to some degree. Second, animal models of TBI have documented fluctuations in DA levels,
11
dysregulation of catecholamine systems, and temporal variation in tissue DA level with change in metabolism in the striatum and frontal cortex in the acute and subacute stages.
12
–14
Third, the neuroprotective and therapeutic strategies that focus on the dopaminergic (DAergic) system have revealed even greater benefits than those that focus on glutamatergic transmission, since strategies that acutely target glutamatergic excitotoxicity to provide neuronal sparing have led to persistent cellular dysfunction even when significant cell sparing occurs.
15,16
In contrast, the benefits of DAergic-targeted strategies are well established in rehabilitative and chronic treatment paradigms.
17,18
Fourth, use of DA agonists have revealed benefits not only in preclinical experiments but also in clinical trials, particularly using chronic administration paradigms; however, there is compelling evidence for targeting DAergic systems also in the acute phase. Moreover, clinical studies have revealed that a DAergic agonist and transporter inhibitors could provide benefits for memory and attention recovery during the chronic and recovery phases.
19
–21
Furthermore, one neuropathology study done by Crane et al.
22
indicated that a single TBI with loss of consciousness is not associated with an increased risk of clinical Alzheimer disease (AD), but late-life effects of TBI may include Lewy bodies, microinfarcts, Parkinson disease (PD), and parkinsonism. Finally, the “posttraumatic stress syndrome (PTSD),” substance abuse, and short-term memory loss after injury may all be related to some degree to deficiency in DA transmission. Thus, this review will focus on the DA system related to brain injury, both acutely and following secondary damage, as follows:
The brain injury model related to the mechanism of injury: How the DA system is affected in different brain injury models The initial and secondary insults related to the DA system Direct insults impact on the DA system Secondary insults alter the DA system
The sequelae of brain injury related to DA transmission impairment
Nigrostriatal pathways
Mesocorticolimbic system
The potential therapeutic value of targeting the DA system after brain injury
Different Brain Injury Models Related to Mechanisms of Injury
Injury from brain trauma can be divided into 2 distinct sequential phases: a primary phase and a secondary phase. The initial, immediate biomechanical damage referred to as primary injury is associated with direct mechanical damage to neurons, supporting cells, and vascular structures. Impact from forces of injury disrupts brain parenchyma and integrity of the blood–brain barrier (BBB). The secondary phase involves subsequent events related to direct insults and is manifested by cascades that impair function, further damage structures, and promote cell death. 23
Different animal injury models result in diverse mechanisms of injury. The most common brain injury animal models include controlled cortical impact (CCI), 24 fluid percussion impact (FPI), weight drop, and explosive (blast) head impact. 23 Each model has unique advantages. The weight-drop model provides biomechanics of injury mechanisms similar to human blunt TBI. FPI and CCI are highly reproducible and allow fine-tuning of injury severity. These are similar to brain contusion injury (Note: Cortical impact has a slightly lower mortality rate but is otherwise similar to fluid percussion.) Blast injury models can provide pathophysiologic and biomechanical mechanisms of explosive injury similar to military TBI settings. 23 The disadvantages of each model include requirement for craniotomy (except for blast) and a need for further standardization.
Alterations in monoamine signaling have been documented after experimental TBI in each of these models. 25 –28 Weight-drop injury has been shown to induce histologic and neurochemical effects that mimic diffuse brain injury in the rat central noradrenergic (NE) system. The data using weight drop revealed that NE turnover and axonal transport were significantly reduced and this effect remained until 8 wk after injury. Therefore, one of the mechanisms of sustained behavioral and psychological abnormalities observed in TBI patients may be chronic suppression of NE turnover. 29 Injury induced by CCI may involve focal changes that could have biomechanical effects similar to focal injury in patients. 30,31 Likewise, blast-related mild TBI (mTBI) may be associated with overpressure waves that produce neuropathological changes similar to diffuse axonal injury. 32 In blast injury animal models, brain regions like the caudate nucleus that were vulnerable to diffuse axonal injury resulting from blast overpressure showed atrophy, which could be one of the mechanisms of working memory loss and posttraumatic stress disorder (PTSD) symptoms. 33 Since different models of injury produce various biomechanical and pathophysiological changes, it is important to understand how these injury models mimic actual brain injury in patients, particularly the differences between the actual and experimental injury models, in order to most effectively translate basic research findings to the clinical arena. Different methods of energy transmission to the brain elicit distinct changes in each animal model: focal or diffuse injury resulting in tissue swelling, increasing intracranial pressure (ICP), BBB breakdown, ischemia, excitatory transmitter release, calcium influx, and then induction of secondary cascades. Nevertheless, the final results or sequelae of TBI may be correlated with neuron loss, axonal injury, and impaired synaptic plasticity. 2
How does the primary injury impact neurotransmission in the DA system? The mechanical damage to neuronal circuits (nigrostriatal or mesolimbic pathways) and the widespread disruption of neuronal projections have implications for all neurotransmitter systems, including DAergic. 11 The vulnerability of the DA system may be related to the long axonal projections in nigrostriatal and mesolimbic pathways, which are easily injured by acceleration and deceleration shearing force during injury. Moreover, results from animal studies show extensive excitotoxic damage, with increasing amounts of oxidative stress induced by TBI in the central nervous system. 34 These primary insults from TBI elicit axon damage, degeneration, and disconnection of postsynaptic neurons following TBI. Therefore, neurotransmission can be severely affected by direct impact after TBI. In addition, the reduction in retrograde transport of neurotrophic factors can also lead to degeneration of neurons via apoptosis or programmed cell death. 35 Brain injury can induce significant DAergic neuronal loss as shown by using tyrosine hydroxylase (TH) immunostaining quantification and retrograde FluoroGold labeling in the substantia nigra (SN) weeks after injury, 36 likely a result of significant mitochondrial dysfunction induced by TBI. 37 The different animal model–induced disruptive mechanisms in the DA system are summarized in Table 1.
The Effect on Dopamine Release in Different TBI Animal Model.

Note: ELISA, enzyme-linked immunosorbent assa; HPLC, high performance liquid chromatography; HVA, homovanillic acid; NA, not applicable; SN, substantia nigra; TH, tyrosine hydroxylase; TBI, traumatic brain injury; VTA, ventral tegmental area.
Secondary Insults after TBI (Related to Dopamine Suppression)
As mentioned above, the pathology of TBI reflects an initial insult, resulting from mechanical damage to neural and vascular structures (primary injury), and the progress of a cascade of subsequent events that impair function, damage structures, and promote further cell death (secondary injury). These secondary pathological processes initially elicited by the primary mechanical changes continue to evolve over a period of days, weeks, or even months after the primary insult. These processes can be mediated by peripheral immune cells and activation of resident brain cells, which trigger cytokine, growth factor, and adhesion molecule release and activate a complex network of pathways. Moreover, neurons are impacted not only by alterations of neurotransmitter release but also by changes in neurotrophic factors (e.g., brain-derived neurotrophic factor [BDNF]) that are critical for their survival. 35 In addition to inflammatory reactions, secondary injury effects involve repair processes, metabolic dysregulation, and apoptosis of neurons and glia. Neurons undergoing apoptosis have been identified not only within the contusion area in the acute posttraumatic phase but also in regions remote from the site of injury in the days and weeks following trauma.
Epigenetic changes following TBI include DNA methylation, chromatin posttranslational modification, and micro ribonucleic acid (miRNA) regulation of gene expression variation. These epigenetic changes and mechanisms might not only be limited to the cell nucleus but also impact the function of mitochondria and neuron recovery processes. 1 Metabolism of essential amino acids can alter gene expression after TBI, which is one potential mechanism of secondary injury. For example, ethionine metabolism provides methyl groups to DNA and histone methyltransferases to epigenetically regulate gene expression, and TBI can alter the methionine metabolism to affect cellular function in multiple organs and systems. 38
Epigenetic changes following TBI may have implications for outcome, 1 recovery, and therapy of TBI 39 ; they can lead to devastating neurological disabilities that impair cognition, memory, movement, sensation, or emotional function. 40 Enormous heterogeneity exists within TBI, and it depends on the severity, location, and whether the injury was focal or diffuse. These epigenetic changes might not only be limited to the cell nucleus but also impact the mitochondria, and such changes have important implications with regard to TBI recovery. 1 However, predicting outcome following a TBI is challenging and cannot be made based solely on clinical presentation and radiological findings, since patients with comparable injuries may have variable outcomes. Therefore, the post-TBI genetic influences on outcome have attracted recent attention. Many genetic variations have been documented after TBI, including changes in neurotrophic factors, cytokines, nitric oxide (NO) syntheses, and tumor proteins such as TP53. Alterations in gene transcription for pathways related to DA transmission after TBI have also been reported. 41,42 For DA neurotransmission, variants linked to DA D2 receptor (DRD2) and ankyrin repeat and kinase domain (ANKK1) genes were found in some individuals with different cognitive recoveries following TBI. Therefore, genetic variation in DRD2 and influences on post-TBI DA transmission may have important implications for cognitive recovery after TBI. 43
The role of genetic factors in the interindividual variability observed in TBI has been examined in predicting functional and cognitive outcome following brain injury. 44 –46 Therefore, not only gene expression alterations associated with mild and repetitive TBI in combat veterans and professional athletes have been studied 47,48 but also numerous genes have been implicated in the pathophysiology and outcome following moderate to severe TBI. 49 A growing body of literature has been devoted to genes involved in TBI, including those that influence the extent of the injury (e.g., pro- and anti-inflammatory cytokine genes), those that affect repair and plasticity (e.g., neurotrophic genes), and those that affect pre- and postinjury cognitive and neurobehavioral capacity (e.g., catecholamine genes). 50 In addition to single-nucleotide polymorphisms that reside in coding sequences and influence expression and protein production, gene expression may be altered without altering the DNA sequence by means of DNA methylation and chromatin modifications. Moreover, the role of epigenetic mechanisms in injury has been explored. 51 For example, genes with biological function clustered to immune responses were significantly upregulated at 4 days after injury, but not at 1 day. 52
The catechol-O-methyltransferase (COMT) gene is believed to functionally modulate DAergic (DAergic) neurons and likely influence executive function. Poorer executive function in TBI patients is thought to be related to decreases in cortical DA, which may be correlated with COMT activity and DA levels. Cheng et al. 28 indicated that the Val variant was associated with perseveration, suggestive of problems with cognitive flexibility, and these authors also claimed an association between the Val allele and response latency following mTBI. 53
Mitochondrial dysfunction following TBI has been well documented in both animal and human studies, with decreased adenosine triphosphate (ATP) production, an increase in lactate accumulation, and dysregulation of intracellular calcium homeostasis prior to neuronal loss. Bulstrode et al. examined the relationship between mitochondrial genomic variants and the 6-mo outcome following TBI; they indicated a significant predictive effect of mitochondrial (mt)DNA genotype on outcome at the 6-mo Glasgow Outcome Scale, when haplogroups were examined individually. 54 Other neurogenerative disorders related to TBI also may involve mitochondrial genes. Oxidative metabolism dysfunction plays a role in AD, while apolipoprotein E has provided allele-specific (E2>E3>E4) neuroprotective effects from oxidative stress in an in vitro study. 55 Mitochondrial dysfunction may also be part of the neurodegenerative diseases’ etiology, especially PD. 56 It has been shown that mitochondrial dysfunction is related to the pathophysiology of PD. Respiratory complex I dysfunction in mitochondria has been found to be reduced in SN of PD, 57 and sporadic PD is characterized by decreased complex I activity. 58 In addition, the increasing complex I catalytic subunits due to excessive oxidative damage of complex I subunits and dysfunction were found in parkinsonian brain. 59,60 Moreover, inherited forms of PD have been linked to mutations in at least 5 genes including: Parkin, 61,62 phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1), 63,64 protein deglycase DJ-1 (DJ-1), 65,66 α-synuclein, 67,68 and leucine-rich repeat serine/threonine protein kinase 1, 69 all of which causally involve mitochondrial dysfunction and oxidative stress in the pathogenesis of PD. Thus, manipulation of mitochondrial genes alters selective vulnerability of SN DAergic neurons, which mimics the clinical degenerative process. 70 Therefore, the damage or suppression of mitochondria function following TBI may also be involved in the altered DA transmission etiology after head injury.
Persistent cognitive deficits after TBI may result from altered DA activity in TBI. The pathophysiology of DA alterations after TBI has been extensively investigated in animal studies. The primary insult may induce axonal injury, with Wallerian degeneration, and structural damage, with BBB breakdown and tissue edema. Secondary injury includes ischemia, excitotoxicity, neuroinflammatory responses, and sequelae of epigenetic and/or genetic expression changes (Fig. 1). 71

The primary and secondary insults from traumatic brain injury (TBI) to dopamine transmission. The responses related to secondary injury includes ischemia, excitotoxicity, neuroinflammatory responses, and sequelae of epigenetic and/or genetic expression changes. AQP4, aquaporin-4; BBB, blood-brain barrier; BH4, tetrahydrobiopterin; DAT, dopamine transporter; EAA, excitatory amino acid; IDO, indoleamine 2,3 dioxygenase; ROS, reactive oxygen species; SNP, single nucleotide polymorphism; VMAT2, vesicular monoamine transporter 2.
Changes in DA transporter (DAT) expression have been shown in the midbrain following TBI. Investigation of DAT levels in the rat frontal cortex and striatum after a CCI revealed that the expression of DAT protein decreased at 2 wk after injury. Because DAT plays an important role in maintenance of DA homeostasis, the decreased expression of DAT after TBI may also result in altered DA neurotransmission in the brain. 72
TBI-induced DA neurotransmission deficits were also attributable to deficits in TH activity. TBI reduces striatal TH activity and potassium-evoked DA release in rats at 1 wk after injury. 73 Subsequent compensatory responses of DAergic neurons upregulate their synthesizing capacity, and a delayed increase in TH expression in rat nigrostriatal system and an efficiency of DA neurotransmission after TBI have been found. 74,75 Therefore, TBI results in functional deficits in DAT expression and transcriptional changes affecting DA protein function that might be improved by treatment with agents, such as methylphenidate, that induce alterations in neurotransmission and enhancement of striatal DA. 76
Striatal function largely depends on DA receptor activation within spiny neurons where the signal transduction cascade involves DA- and 3′,5′-cyclic adenosine monophosphate (cAMP)–regulated phosphorylation of 32 kDa (DARPP-32) and protein phosphorylation 1 activity. For example, a CCI animal study revealed that TBI decreased DARPP-32 phosphorylation and reduced protein phosphorylation 1 activity, resulting in striatal dysfunction. 77 On the other hand, in vivo stimulation of the median forebrain bundle evokes DA release in striatum and this release was suppressed after CCI (TBI), while DAT expression decreased proportionally in injured striatum as well. 25
TBI in adult rats causes progressive nigrostriatal DAergic cell loss related to increased microglial activation in the SN and has been shown to cause loss of up to 15% of DAergic neurons after ipsilateral TBI. At the chronic stage (26 wk postinjury), TBI induces a loss of up to 30% DAergic neurons bilaterally, which suggests that TBI is able to induce progressive degeneration of nigrostriatal DAergic neurons. 78 Pharmacotherapies targeting DA (DA) have shown benefits in attention, behavioral outcome, executive function, and memory. 79
Secondary Insults Related to the Dopamine System
Neuroinflammation as the result of injury
The activation of microglia is a key step in neuroinflammation (Fig. 2). In an FPI model, the activation of microglia was found not only in the ipsilateral but also in the contralateral striatum, distant from the impaction side, 71 whereas microglia were scanty in control animal striatum (A contralateral and B ipsilateral side). Microglia increased after a 2- or 6-psi FPI from acute stages (1 day postinjury) and subacute stages (1 wk postinjury) and persisted until chronic stages (8 wk postinjury) especially in a 6-psi injured group.
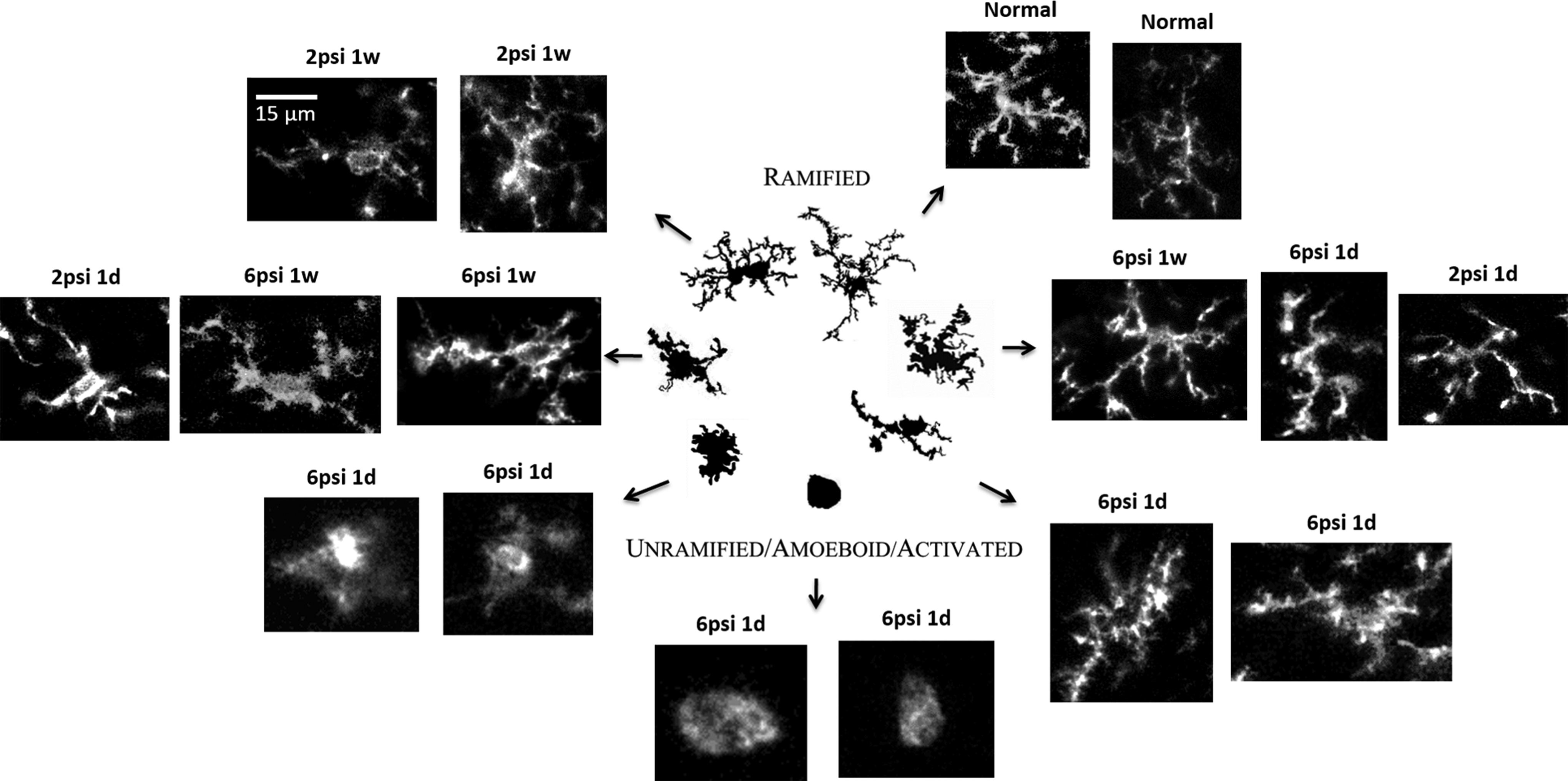
The morphology of microglial changes after traumatic brain injury (TBI); the morphology of each stage is related to their reversible morphologies from a simple rounded to complex branched forms (the figure was modified by adding our unpublished histological data on microglia after TBI, and the central illustration panel was adapted from Karperien et al. 124 psi: pounds per square inch (lbf/in) is a unit of pressure).
In FPI animals, both tonic and bursting DA release, reuptake and release probability were suppressed on both the ipsilateral and contralateral sides of the striatum from the acute to chronic stages. These neuronal parameters were also affected in the subacute stage on both sides of the striatum in 2-psi injured animals, and the turnover rate of DA was significantly altered. TBI not only suppresses DA release and reuptake but also affects DA metabolic rate and release probability, and these effects are found both ipsilateral and contralateral to the injury during both the acute and subacute stages after injury. 71
Not only the nigrostriatal but also the mesolimbic system is affected by TBI. Persistent neuroinflammatory responses have been observed in the cerebral cortex, nucleus accumbens (NAC), and ventral tegmental area (VTA) after CCI, with significant increases in astrocytic glial fibrillary acidic protein and microglial ionization basic acid 1 markers in the NAC at the conclusion of conditioned place preference (CPP) testing. In both the cortex and NAC, numerous inflammatory genes were upregulated in moderate CCI-TBI animals. TBI-induced chronic mesolimbic neuroinflammation may thus contribute to the long-term changes in rewarding effects in adulthood. 80
After TBI, amyloid precursor protein (APP) accumulates in white matter tracts, and this leads to deficits in striatal DARPP-32 phosphorylation. These downstream signaling mediators of striatal DAergic neurotransmission increased sensitivity to ethanol-induced sedation while altering ethanol consumption. 81 Moreover, involvement of the nucleus accumbens in depression after mTBI may suggest an underlying dysfunctional reward circuit that affects the prognosis in these patients. 82 Using diffusion tensor imaging (DTI) to investigate depression after TBI 69 and volumetric analysis of the ventral striatum in adults with TBI, it has been shown that the functional deficits in reward and cognition are related to damage to the ventral striatum and its associated structures. 70
Epigenetic changes after TBI Related to DA System
Epigenetic alterations after TBI have been shown in several different TBI animal models, with variations in DNA methylation, histone acetylation, miRNA, and altered inflammatory responses resulting in neuroinflammation. For example, blast-induced neurotrauma (BINT) can alter epigenetic transcriptional regulation through DNA methylation, 83,84 which induces imbalance between DNA methylation and demethylation and leads to altered methylation patterns and subsequent changes in gene transcription. These BINT-induced gene expression abnormalities may involve oxidative stress, inflammation, and hippocampal neuronal apoptosis. There were significant negative correlations between global DNA methylation and the magnitude of blast overpressure. These data offer insights into TBI outcomes associated with pathology including inflammation, oxidative stress, and apoptosis. 83 Moreover, genome-based evidence has indicated that multiple blast overpressure exposures could induce changes in DNA methylation for genes involved in converting serotonin to the circadian hormone melatonin. This could result in sleep disturbances and depression associated with TBI. 85 TBI may also induce inflammation that produces mitochondrial dysfunction and oxidative stress, which play synergistic roles in DAergic neurodegeneration in the nigrostriatal system. 86 Attenuation of neuroinflammation could promote DAergic neuronal survival in the nigrostriatal system of rats after diffuse brain injury. 36
The Sequelae of Brain Injury Related to the Dopamine Transmission Impairment
TBI-related nigrostriatal pathway impairment
TBI via FPI induces suppression of the field postsynaptic potentials and an imbalance of excitatory and inhibitory synaptic activity in the prefrontal cortex, evidenced by in vitro electrophysiology and alterations in working memory. 87 TBI often results in multiple neuropsychiatric sequelae, including cognitive, emotional, and behavioral problems, which have been correlated with lower DA concentrations in the striatum of the injured rats 25,75 as well as with the degeneration and apoptosis of DAergic neurons in the SN. 88 Amantadine (AMT) therapy restores DA levels in the striatum, decreases degeneration and apoptosis of DAergic neurons in the SN, and significantly ameliorates depression-like behavior. 89
Following injury, pro-inflammatory cytokines and chemokines are acutely increased, which results in microglial activation and damage to DAergic axons in the striatum and SN during the acute stage of TBI. These deleterious effects are ameliorated by pioglitazone. 90 Significant overexpression and abnormal accumulation of α-synuclein could be found in inflammation-infiltrated SN, as well as significant decreases in TH-positive expression in surviving DAergic neurons of the SN pars compacta of rats exposed to chronic TBI. 88
To study remote effects on the striatal DA system after fluid percussion injury, microglial reactions were analyzed using immunohistochemistry at multiple stages after injury. In severe fluid percussion injury, TBI suppresses DA release and reuptake and affects the metabolic rate and release probability of DA on both sides of the nigrostriatal system, ipsilateral and contralateral to the injury, during both the acute and subacute stages after the injury. 71 The chronic stage after TBI may also affect injured nigrostriatal or mesolimbic system neuronal regrowth based on chondroitin sulfate (CS) and dermatan sulfate (DS) in the lesion scar that impedes axonal regeneration after traumatic injury in the mouse brain. One animal study demonstrated that DS and CS play different functions after brain injury; DS is involved in the lesion scar formation, and CS inhibits axonal regeneration. 91
Parkin protects DAergic neurons from oxidative stress–mediated death by regulating mitochondrial function. TBI in Parkin null mice induces profound injury and cell death in the central nervous system due to oxidative stress. This leads to lipid peroxidation and protein and nucleic acid oxidation, which play a significant role in secondary injury. 92 With data from molecular mechanisms of secondary injury after TBI, therapeutic strategies for long-term therapy have been gradually updated. Evidence for neuroprotective effects was provided for pituitary adenylate cyclase activating polypeptide (PACAP27) in mouse models of PD, which involved the modulation of potassium (ATP) subunits and D2 receptors in the striatum. 93 Furthermore, TBI may exacerbate nigrostriatal DAergic degeneration by modulating PD-associated genes like α-synuclein, which is suspected to be a pathological link and therapeutic target in both PD and TBI. 94 A delayed increase in TH in the nigrostriatal system after TBI during the chronic state may suggest that a compensatory upregulation of DA synthesis capacity and an elevated DA transmission exist in the recovery stage of TBI. 74
TBI-related mesolimbic system and prefrontal cortex dysfunction
Psychiatric disorders like depression, anxiety, and substance abuse are prevalent after TBI. Using the CCI model of TBI and the CPP assay, neuroinflammatory responses have been observed in the cerebral cortex, nucleus accumbens (NAC), and VTA after CCI. 80 These results suggest that sustaining moderate TBI during adolescence might generate long-term enhancement of rewarding effects of psychostimulants in adulthood and that TBI-induced chronic mesolimbic neuroinflammation plays a crucial role in this process. 80 An immunohistochemical and in situ hybridization study showed that DAT expression changed in the midbrain following TBI. DAT plays an important role in recycling DA (DA) from the synapse and maintaining DA homeostasis, so the decreased expression of DAT after TBI may result in altered DA neurotransmission in the brain. 72
Neural plasticity and neurogenesis effects of environmental enrichment (EE) in neurodegenerative diseases like PD have been studied. 95,96 The restoration of monoamine (DA and serotonin) transporter function after EE has also been documented. 97 Thus, the effect of EE on acute neuronal injury like TBI has attracted some attention. TBI elicited significant alterations in DAergic systems and in important genes for signal transduction, calcium signaling pathways, membrane homeostasis, and metabolism in SN and VTA. 72,98 These changes are modulated by EE after TBI. Therefore, the potential roles of EE in TBI rehabilitation are indicated. 98
A systematic review of the clinical literature indicates that no single pharmacological therapy will unequivocally improve clinical outcomes after TBI. Five agents have demonstrated promising clinical benefits for specific TBI patients: statins, N-acetyl cysteine, enzogenol, cerebrolysin, and NO synthase inhibitor (VAS203). Statin treatment could markedly reduce functional neurological deficits after TBI in mice, and this is accompanied by reduction in degenerating hippocampal neurons and suppression of inflammatory cytokine mRNA expression in brain parenchyma and improved cerebral hemodynamics following head injury.
99,100
The neurorestorative mechanism of statins may be mediated through activation of the protein kinase B–mediated signaling pathway, subsequently upregulating expression of growth factors (e.g., BDNF)
101
and possibly via activation of Notch signaling to induce neurogenesis in the dentate gyrus of the hippocampus.
102
This can lead to restoration of cognitive function after TBI in rats.
103
NAC effects on TBI are controversial, with some studies showing extended behavioral recovery after injury.
104,105
Enzogenol is well tolerated and may reduce self-perceived cognitive failures in patients 3 to 12 mo post-mTBI
106
and can improve clinical outcomes after TBI.
105
Cerebrolysin is a parenterally administered neuropeptide preparation that can penetrate the BBB and can act in a manner similar to endogenous neurotrophic factors. Cerebrolysin could decrease brain APP accumulation and astrogliosis as well as increase neuroblasts and neurogenesis to improve long-term cognitive function. It elicits neuroprotection and neural recovery after TBI using a multidimensional ensemble of outcome scales in mTBI.
107
–110
NO has previously been shown to be involved in damage of the BBB; the NO synthase inhibitor (VAS203) showed the same vasoconstrictive effect as the classical NO synthase inhibitor
Dysregulation in both inhibitory and excitatory neurotransmission by alterations in cytosolic protein complexin levels has been shown after TBI. Increasing excitatory neurotransmission initially may increase oxidative stress, which plays an important role in neuronal cell loss following TBI. This may contribute to the pathophysiology of cerebral damage following brain injury that is responsive to antioxidant treatments such as N-acetyl cysteine. 104,113
Even mTBI induces changes in immunoreactivity for inducible transcription factors (e.g., c-Fos, c-Jun, JunB and Krox-24, etc.) in areas related to fear condition (e.g., amygdala), reward effects, and working memory (e.g., hippocampus, striatum, NAC, etc.), which are distal to the injury site. 114 Differences in neural connectivity between the SN and VTA in the human brain revealed by DTI have showed that connectivity of SN was higher than that of the VTA. Therefore, differences in neural connectivity between the SN, VTA, and other areas in the human brain may also underlie differential vulnerability of the DA system versus other regions. 115
The Therapeutic Value of Targeting the Dopamine System after Brain Injury
AMT blocks N-methyl-
Treatment with low-dose methamphetamine improves behavioral and cognitive function after severe TBI and is able to restore learning and memory function to near normal levels after TBI.
118
Oxidative stress is another significant contributor to the secondary sequelae of TBI and may mediate subsequent neurobehavioral deficits and histopathology. Bromocriptine, a DRD2 agonist with significant antioxidant properties, enhances spatial learning and hippocampal neuron survival in a rodent model of focal brain trauma
119
as well as working memory in a CCI animal model.
120
Moreover, results from pharmacological restoration of DA transmission using postinjury administration of
However, side effects of DA treatment in TBI should also be considered. Contrasting effects of DA therapy have been shown in brain injury animal models. Dopamine was capable of restoring CPP in a model of rapidly rising ICP, but there was only partial restoration of CBF. Therefore, although DA is capable of partially restoring CBF after injury, worsening of the brain swelling processes subsequently due to the effects of vasopressor therapy in a clinical setting needs to be more carefully evaluated. 122
Using deep brain stimulation implantation in chronic, severe TBI patients to bilaterally stimulate NAC and anterior limb of the internal capsule could modulate the prefrontal cortex and improve independence in self-care and activities of daily living. 123 However, this is another approach that must be confirmed and further evaluated.
In conclusion, there are many molecular and cellular changes in TBI, with no clear-cut therapies available. Elucidation of the cellular and molecular changes may lead to novel therapeutic approaches to treat this “silent” epidemic.
Footnotes
Author Contributions
Yuan-Hao Chen and Eagle Yi-Kung Huang provided equal contribution.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by the National Science Council of Taiwan under grant NSC102-2314-B-016-030∼MY3, NSC 105-2314-B-016-001-MY3, and by Medical Research Project grants TSGH-C105-075, TSGH-C103-083, TSGH-C104-187 from the Tri-Service General Hospital of Taiwan and MAB-105-105 from National Defense Medical Center. This project was also supported in part by philanthropic support from the George R. and Constance P. Lincoln family.