
Abstract
Regulatory lymphocytes play a pivotal role in preventing organ-specific autoimmune disease and in induction and maintenance of tolerance in various experimental transplantation models. The enhancement of the number and activity of peripheral CD4+CD25+ Treg cells is an obvious goal for the treatment of autoimmunity and for the suppression of alloreactions. The present study demonstrates that naturally occurring CD4+CD25+ Treg (nTreg) cells preferentially proliferate to a fourfold increase within 3 days in response to the administration of a single superagonistic CD28-specific monoclonal antibody (supCD28 mAb). The appearance of increased Foxp3 molecules was accompanied with polarization toward a Th2 cytokine profile with decreased production of IFN-γ and increased production of IL-4 and IL-10 in the expanded Treg subset. Adoptive transfer of supCD28 mAb-expanded cells in a graft-versus-host disease (GvHD) model induced a potent inhibition of lethality. These results suggest that this therapeutic effect is mediated by the in vivo expansion of nTreg cells. Taken together, these data demonstrate that supCD28-mAb may target nTreg cells in vivo and maintain and enhance their potent regulatory functions for the treatment GvHD.
Introduction
CD4+CD25+ regulatory T (Treg) cells play a central role in the suppression of autoimmunity, inflammation, and allograft rejection (1,6,11,12). Therefore, therapeutic agents that are capable of enhancing the number and activity of this T-cell subset are highly desirable. However, the normal physiology of this population of suppressor T cells is still not completely understood and the molecular basis for the anergic state of CD25+ T cells remains only partially known. A crucial area for future studies is the identification of drugs, cytokines, or costimulatory molecules that reverse anergy and enhance growth, but preserve the suppressor function of the CD4+CD25+ Treg cell population.
A unique superagonistic CD28-specific monoclonal antibody (supCD28 mAb) was developed to induce proliferation of all primary resting T cells without TCR engagement, which potently stimulates both the in vivo and vitro expansion of CD4+CD25+ Treg cells (16,26). T-cell activation by supCD28 mAb occurs without an increase in ZAP-70 and TCR phosphorylation and hence without stimulation of the TCR complex (17). In keeping with the Th2-promoting effect of CD28 signals in costimulation, it was shown that T-cell activation with supCD28 primed CD4+ T cells for Th2 differentiation in vitro and induced IL-4 and IL-10 expression along with Th2-dependent Ig isotypes in vivo (21). Recently, studies in rodent autoimmune disease models have shown the efficacy of supCD28 mAb treatment in prevention of experimental autoimmune neuritis, experimental autoimmune encephalomyelitis with a marked reduction of infiltrating cells and IFN-γ production (23), adjuvant arthritis with an amelioration of Th1/Th2 cell balance and the induction of high IL-10 expression in synovia (20), and in a type 1 diabetes BB rat with an increase of CD4+CD25+ T-cell frequency (2).
The present study showed that in vivo application of the supCD28 mAb to Lewis rats causes preferential expansion of Foxp3-expressing Treg cells over conventional T cells and maintains not only their phenotype but also their potent regulatory functions against T-cell proliferation in response to mitogens, antibodies, and allostimulation in vitro. Furthermore, CD28-activated Treg cells suppress the alloreactive T-cell response in a model of graft-versus-host disease (GvHD) in vivo, thus indicating that this therapeutic effect is mediated by the in vivo expansion of naturally occurring Treg (nTreg) cells.
Materials and Methods
Animals
Adult male Lewis (RT1l), DA (RT1a), and F1 (Lewis × DA) rats were purchased from Shizuoka Laboratory Animal Center (Shizuoka, Japan) and used at 4–6 weeks of age. The animals were maintained under standard conditions and fed rodent food and water according to the laboratory animal care principles and the guide for the care and use of laboratory animals established by our institution.
Total Spleen T-Cell Purification
For the preparation of purified spleen T cells, supCD28 mAb-treated (1 mg/rat, IV) or mIgG-treated control Lewis rat spleens were harvested and gently ground with frosted objective slides in PBS. They were filtered through 200-μm nylon mesh for single-cell suspension preparation and then separated by Lympholyte-Rat (Cedarlane Laboratories, Ontario, Canada). The splenocytes were washed two times after lysing the erythrocytes with RBC lysis buffer (55 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). The purified T cells were then prepared through a nylon column (Wako Pure Chemical Industries, Ltd. Osaka, Japan) for in vitro culture in GIT medium containing 50 mg/L kanamycin sulfate (NIHON Pharmaceutical, Tokyo, Japan) supplemented with 50 mM 2-mercaptoethanol (Wako Pure Chemical Industries, Ltd.).
CD4+CD25+ and CD4+CD25- T-Cell Purification
For the isolation of purified CD4+CD25+ and CD4+ CD25- T cells, splenocytes were first stained with FITC-conjugated anti-rat CD8, CD11b/c, and NK1.1 (BD Pharmingen, San Diego, CA) followed by anti-FITC MicroBeads (Miltenyi Biotec, K.K. Tokyo, Japan) and anti-rat Kappa MicroBeads (Miltenyi Biotec) and than sorted on an AutoMACS system (Miltenyi Biotec). To isolate CD4+CD25+ T cells, the negative fraction of the enriched CD4+CD8- cells was stained with biotinylated anti-rat CD25 (BD Pharmingen) followed by antibiotin MicroBeads (Miltenyi Biotec) and sorted on an Auto MACS. The negative fraction, enriched CD4+CD25- cells, was labeled with anti-rat CD4 MicroBeads (BD Pharmingen) sorted on an Auto MACS. The enriched T-cell populations were evaluated by FACScan (Becton Dickinson) and more than 98% purity of the CD4+ CD25+ and 95% purity of the CD4+CD25- cells population were routinely obtained.
Quantitative Estimation of Mitotic Events and Precursor Frequencies by Cell Tracking Dye
The cells were resuspended at a density of 1–2 × 107 cells/ml in GIT. An equal volume of 15 mM of carboxyfluorescein diacetate-succinimidyl ester (CFSE, Molecular Probes Inc., Eugene, OR) in GIT was added and then the cells were cultured at 37°C for 10 min. The reaction was quenched by the addition of 10 times volumes of cold GIT medium. Labeled cells were washed twice before use in cold medium. SupCD28 mAb-treated T cells were prepared alone or by an equal number mixed with CFSE-labeled naive purified T cells (5 × 105) and cultured with 20 Gy γ-irradiated DA splenocytes (APC cells, 5 × 105) or in the presence of ConA (2 mg/ml; Wako, Ltd.) in a 24-well plate. After 3 days, the cells were harvested from the plate and washed with PBS. Flow cytometric analysis was performed using a Becton Dickinson Immunocytometry System and a FACS Calibur cytometer (San Jose, CA). Dead cells were excluded from the analysis by staining with propidium iodide. Division peaks were determined by measuring the histogram fluorescence intensity of the CFSE. Quantitative estimation of mitotic events was achieved as described previously (7). Briefly, the number of progenitors required to give rise to daughter cells at each division peak (as determined by CFSE intensity) decreases exponentially. This may be expressed using the equation 1/2n, where n is equal to the number of divisions that cells have undergone in relation to the number of undivided cells (represented by n = 0). The precursor frequency is then calculated by dividing the total number of precursor cells giving rise to progeny by the total number of cells. The absolute number of mitotic events is derived by subtracting the number of division precursors from the number of daughters generated by each precursor population. The sum of these events represents the total number of cell divisions that have occurred in the labeled cell subset by the time of harvest.
T-Cell Proliferation Assays
CD4+CD25 T cells from supCD28 mAb-treated or mIgG-treated control Lewis rat spleens were prepared by serial dilution (1:0.25, 1:0.5, and 1:1) mixed with CD4+CD25- T cells (1 × 105) and cultured with 20 Gy γ-irradiated DA splenocytes (APC cells, 1 × 105) in a U-bottom 96-well plate (IWAKI, Tokyo, Japan) at a final volume of 200 μl/well in humidified atmosphere at 37°C for 5 days. The proliferation of T cells was measured with cell proliferation ELISA kits (Roche Diagnostics Gmbh, Penzberg, Germany) (9). Briefly, the cells were labeled with 5-bromo-2-deoxyuridine (BrdU) solution at 10 μl/well and incubated for an additional 2 h at 37°C. After centrifugation, the supernatant was removed, 200 μl/well of fixation and DNA denaturation (FixDenat) solution was added to the cells and they were reincubated for 30 min at 15–25°C. The cells were cultured for 90 min with anti-BrdU-peroxidase solution and subsequently washed three times. After adding substrate solution at 100 μl/well, the BrdU incorporation was measured with a chemiluminescence reader (Wallac ARVOTM SX; PerkinElmer, Inc., Wellesley, MA) and the data were processed using Wallac1420 manager software (PerkinElmer).
Flow Cytometric Analysis
CD4+CD25+ and CD4+CD25- T cells from supCD28 mAb-treated or IgG-treated control Lewis rat spleens were collected and suspended in PBS and incubated at 4°C for 30 min with an optimal concentration of Cy-Chrome conjugated anti-rat CD4 antibody (OX-35, BD Pharmingen) in combination with FITC-conjugated anti-rat CD25 antibody (OX-39, BD Pharmingen) diluted with PBS containing 1% fetal calf serum. The membranes were stained and intracellular staining was then performed using a BD Cytofix/Cytoperm Kit (BD Pharmingen). The cells were fixed with 100 μl of BD Cytofix/Cytoperm™ (BD Pharmingen) at 4°C for 20 min. The cells were next washed twice with BD Perm/Wash™ buffer (BD Pharmingen) and then resuspended in 50 ml of the same buffer containing the optimal concentration of PE-conjugated anti-Foxp3 antibody (FJK-16s, eBioscience, San Diego, CA) for permeabilization. The cells were gently mixed and incubated for 30 min in the dark at 4°C. They were then washed twice with BD Perm/Wash™ buffer and resuspended in a staining buffer. Thereafter, the cells were analyzed by flow cytometry (FACScan, Becton Dickinson).
RNA Preparation and Semiquantitative RT-PCR
The total cellular RNA was isolated from the CD4+ CD25+ and CD4+CD25- T cells from supCD28 mAb-treated or mIgG-treated control Lewis rat spleens using the RNeasy Mini Kit (Qiagen Inc., Valencia, CA) according to the manufacturer's protocol (8,14). The concentration of total RNA was determined by measuring the optical density at 260 nm. The quality of the total RNA was checked by using an Agilent 2100 Bioanalyzer and RNA 6000 Nano LabChip® Kit (Agilent Technologies, Palo Alto, CA) according to the manufacturer's protocol. RNA was converted to cDNA using Super Script™ III (Invitrogen, Carlsbad, CA) for RT-PCR. PCR primers were designed on the basis of the reported cDNA sequences; the expected fragment sizes for these genes are specified in Table 1. The cDNA fragments for each gene were amplified in optimized cycles (94°C for 30 s, 60°C for 30 s, and 72°C for 30 s), followed by a 10-min extension at 72°C by a DNA thermal cycler (PerkinElmer). The amounts of the PCR products were detected by electrophoresis in 1.8% agarose gel in a TBE buffer. The electrophoresis images of the most quantitative cycle points of PCR were used to evaluate the expression levels of the genes by ScionImage (Scion Corp., Frederick, MD) and aligned the results by the expression levels of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene for comparative analysis.
Oligos Used in This Study

Assay of Local GvHD Reactivity
The popliteal lymph node assay was used according to the modified method as described previously (9). Single-cell suspensions (5 × 106) were prepared from either supCD28 mAb-treated rat or mIgG control rat spleen and subcutaneously injected into the left or right footpad of 4–5-week-old F1 (Lewis × DA) hybrid recipients. Their bilateral popliteal lymph nodes were removed 7 days later and dissected free of fat and surrounding tissue and weighed to within an accuracy of 0.1 mg.
Assay of Systemic GvHD Reactivity
Single-cell suspensions (2.5–5 × 108) were prepared from either supCD28 mAb-treated rat or mIgG control rat spleen and intravenous injected into 4-week-old F1 (Lewis × DA) hybrid recipients. The rats were weighed on alternate days during the active phase of the GvHD. Animals undergoing typical GvHD showed rapid weight loss and commonly described signs of the disease, including ruffled fur, reddening of the skin, a hunched posture, and ultimately death.
Statistical Analysis
Student's t-test was used to compare the paired and unpaired variables. A statistical evaluation for graft survival was performed using the Kaplan-Meier test. Value of p < 0.05 were considered to be statistically significant. All in vitro experiment data were representative of three independent experiments and expressed as the mean ± SD.
Results
SupCD28 mAb-Expanded T Cells Suppress Naive T-Cell Proliferation in Alloantigen-Induced Stimulation
A single injection of the supCD28 mAb JJ316 led to a significant increase of the CD4+CD25+ population in the spleen, peripheral blood lymphocyte (PBL), and lymph nodes from an initial 3–5% to about 15% at 72h after stimulation (Fig. 1A). Figure 1B shows that supCD28 treatment dramatically increased the representation of the CD4+CD25+ population among the CD4 cells of the spleen (4.5-fold), PBL (5.18-fold), and lymph node (LN; 8.15-fold).

SupCD28 mAb-induced CD4+CD25+ T-cell expansion. (A) The proportion of CD4+CD25+ T cells was increased in the spleen, peripheral blood lymphocyte (PBL), and lymph node (LN) cells in supCD28 mAb-treated group compare to the control mIgG-treated group. (B) SupCD28-treated only dramatically increased CD4+CD25+ cell population of the spleen, PBL, and LN. The population of the CD4+CD25- cells had no changes in these two groups. Data are representative of three independent experiments and indicate the mean ratio of triplicate results in each experiment.
Next, the ability of lymphocytes from CD28-stimulated rats to suppress in vitro proliferative CFSE-labeled naive T-cell responses to alloantigens was tested. Without the addition of cells, the quantitative estimation of the absolute number of mitotic events represented by each histogram was 2,520 (allo-APC) (Fig. 2A). Addition of equal numbers of lymphocytes (2.5 × 105/well) from control mouse IgG (mIgG)-treated rats was without effect, whereas the addition of lymphocytes from supCD28 mAb-treated rats resulted in a reduction from 2,520 to 1,403 mitotic events (56%) in the allo-APC-induced responses, respectively. Moreover, the bivariate dot plots showed a significant suppressive effect on the division of the CD4 T-cell subset of the CFSE-labeled lymphocytes by the same stimuli (Fig. 2B).

SupCD28 mAb-expanded lymphocytes suppressed the naive T-cell proliferation via allostimulation. (A) CFSE-labeled naive Lewis lymphocytes were stimulated with allogeneic APCs. The CFSE fluorescence profiles of the live lymphocytes from each culture at 72 h and quantitative estimation of mitotic events represented by each histogram are shown. (B) Bivariate dot plots show the cell division on the CD4 T cell subset of CFSE-labeled lymphocytes stimulated with APC. The data clearly show that the division of the naive lymphocytes was suppressed when mixed at an equal ratio of expanded lymphocytes compare to naive and mIgG groups. Data are representative of three independent experiments.
SupCD28-Expanded CD4+CD25+ T Cells Show Higher Levels of IL-2, -4, -10, Foxp3, and Suppressive Capacity Than Naive nTreg Cells
Next, the expression of Treg-related genes was investigated (3,5,10). The expanded CD4+CD25+ T cells expressed much higher levels of IL-4 and IL-10 mRNA in comparison not only to CD4+CD25- T cells but also to nTreg cells (Fig. 3, upper). These results were also confirmed by an FCM analysis showing that expanded CD4+CD25+ T cells expressed higher levels of intracellular IL-4 and IL-10 (data not shown). With regard to IL-2 mRNA, there was an increased expression in comparison to nTreg cells from unstimulated animals and no significant difference in the IL-2 mRNA expression between the CD4+CD25+ and CD4+CD25- T cells from the supCD28 mAb-stimulated rats. Because CD4+CD25+ cells from supCD28-stimulated animals contain a small but significant fraction of Foxp3-negative, activated effector CD4 T cells, these are the likely source of IL-2 mRNA detected in this population. In addition, purified CD4+CD25- T cells had no expression of Foxp3 after in vivo supCD28 mAb stimulation and both naive and supCD28 mAb-expanded CD4+CD25+ cells exhibited high levels of Foxp3 mRNA (Fig. 3A, lower).

Cytokines, Foxp3 expression, and suppressive capacity of the supCD28-expanded CD4+CD25+ cells in vivo, and CD4+CD25+Foxp3+ Treg cells can be expanded by activation of natural CD4+CD25+ Treg cells via supCD28 mAb, not be converted from CD4+CD25-Foxp3- T cells in vitro. (A) Compared to mIgG control (upper panel), in vivo supCD28-expanded CD4+CD25- and CD25+ T cells expressed significant amounts of IL-2, while CD25+ cells expressed high levels of IL-4 and IL-10 mRNA by semiquantitative RT-PCR assay. Purified CD4+CD25- cells had no expression of the Foxp3 after the in vivo supCD28 mAb stimulation, and both naive and supCD28-expanded CD4+CD25+ cells exhibited a high level of Foxp3 mRNA expression (lower panel). (B) In vivo expanded CD4+CD25+ Treg cells can potentially inhibit the CD25- T-cell proliferation via allostimulation in a dose-dependent manner as well as naive cells (rlu/s: relative light units/second). (C) FACS plots showing Foxp3 expression of the sorted cells in each population before activating (left) and in vitro stimulated supCD28 mAb for 72 h. In vitro supCD28-expanded rat CD4+CD25+ Treg cells remained positive for not only CD25 but also Foxp3 high expression. In contrast, all CD4+CD25- T cells expressed CD25 after activation, but the Foxp3 expression was very low. Data are representative of three independent experiments.
To specifically examine the suppressor cell activity of supCD28-expanded CD4+CD25+ cells, CD4+CD25+ T cells were isolated from naive rats and rats at day 3 of supCD28 stimulation as described above. Suppression assays were performed with increasing numbers of autologous naive or supCD28-expanded CD4+CD25+ T cells to test their suppressive activity using allogeneic APCs. Figure 3B shows that at a ratio of 1:1, CD4+ CD25+ T cells inhibited the proliferation of naive CD4+ CD25- T cells stimulated with alloantigen by allogeneic APCs (75.0 ± 2.9%). These data demonstrate that supCD28 mAb-expanded CD4+CD25+ T cells are even more suppressive than CD4+CD25+ nTreg cell populations from normal naive rats.
Expansion of nTreg Cells by Stimulation of supCD28 mAb Occurs Without Conversion From CD4+CD25- Cells
Lin and Hünig (16) have previously shown that the increased number of CD4+CD25+ cells found in supCD28-stimulated rats are derived from preexisting CD4+CD25+ cells rather than by conversion from CD4+CD25- cells. Here, these experiments were extended to determine in vitro whether upregulation of CD25 on the latter population (which plays little role in vivo) is associated with the induction of Foxp3 expression. As shown in Figure 3C lower panels, in vitro supCD28 mAb stimulation induced expression of CD25 in CD4+CD25- T cells, which contained only a few Foxp3-positive cells (Fig. 3C, upper right), while all supCD28 mAb-expanded rat CD4+CD25+ Treg cells remained positive for not only CD25 but also Foxp3 high expression (Fig. 3C, upper right).
SupCD28 mAb-Expanded T Cells Suppress GvHD Reactions
Next, a clinically relevant in vivo model was used to investigate the role of supCD28 mAb-expanded lymphocytes on alloresponses in vivo and the adoptive transfer of alloreactive T cells that results in local GvH reactivity and systemic GvHD lethality (Fig. 4A). First, a popliteal lymph node assay was performed using F1 (Lewis × DA) hybrid recipients to test the function of supCD28 mAb-treated splenocytes in local GvH reactivity. Control mIgG-treated Lewis splenocytes were injected into the right footpad of the recipients and supCD28 mAb-treated splenocytes into the left pad at the same dose. No enlargement was found in the PLN on the left side, injected with supCD28 mAb-treated splenocytes (8.9 ± 5.7 mg) in comparison with control (40.4 ± 13.4 mg) (Fig. 4B). Secondly, the systemic GvH reactivity of supCD28 mAb-expanded splenocytes was tested by injecting either population IV into F1 (Lewis × DA) hybrid recipients. As shown in Table 2, the rats receiving 5 × 108 supCD28 mAb-expanded splenocytes survived to 45.5 ± 20.3 days and in the 2.5 × 108 transfer group, the supCD28 mAb-expanded spleen T cells did not mediate GvHD lethality in any recipients in comparison to the control (14.3 ± 1.2, 18.3 × 6.5 days; 5 or 2.5 × 108 cells, respectively). Furthermore, cotransfer of equal numbers of supCD28 mAb with naive and expanded spleen cells inhibited the GvHD mortality significantly (41.8 ± 19.3 days) in comparison to recipients receiving only naive spleen T cells (p = 0.0001) (Fig.4C, D). Consistent with these observations, Figure 5A shows that extensive perivascular infiltration of mononuclear cells and tissue damage were seen in histological studies of the skin, tongue, lung, and liver from the control mIgG-treated lymphocytes transferred host on the day 14 (left panels). In contrast, relatively few infiltrating mononuclear cells and almost normal tissue architecture were observed in supCD28 mAb group (right panels). These results indicate that supCD28 mAb-treated lymphocytes have potent immunoregulatory activity on the response to allogeneic MHC antigens, even in a more sensitive assay.

SupCD28 mAb-expanded lymphocytes suppressed GvH reactions. (A) Experiment design. (B) Local GvH assay: the popliteal lymph nodes on the left side of the F1 (Lewis × DA) rat, in which supCD28 mAb-expanded lymphocytes alone were injected, were not enlarged compared with the mIgG-treated control Lewis lymphocytes injected alone on the right side. (C, D) Systemic GvH assay: transfer of the mIgG-control Lewis lymphocytes to F1 rat leads to definite lethality in a nonirradiated rat GvHD model. The host's weight loss and survival rate was dependent on transferred lymphocytes. Nonetheless, expanded lymphocytes alone from supCD28 mAb-treated Lewis rats suppressed the GvH reaction and rats revealed slight weight loss and no animal died in the 2.5 × 108 cells (open boxes) transferred group within observation times of (over 60 days). In addition, the protective effect of the transferred supCD28 mAb-expanded cells (2.5 × 108; open triangles) was still apparent when mixed with an equal ratio of control Lewis lymphocytes.
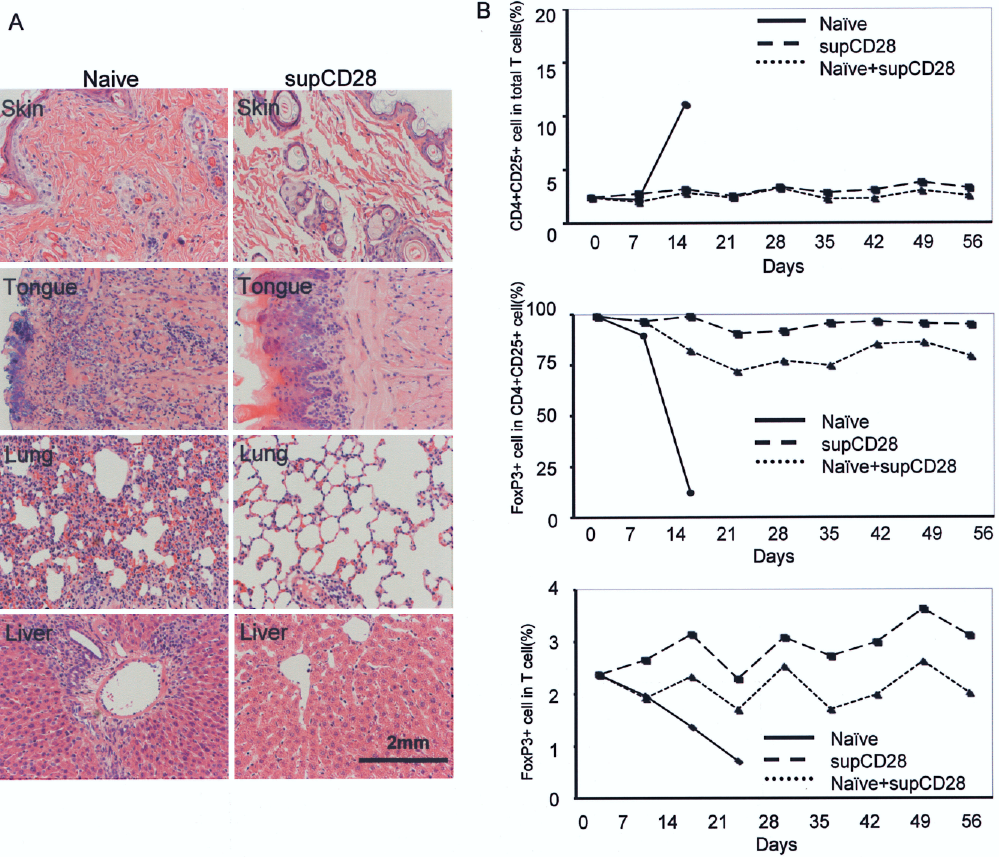
Histological analysis of cell infiltration and tissue injury in GvHD recipients and kinetic changes of the Foxp3 Treg cell populations in host PB. (A) H&E sections of the skin, tongue, lung, and liver from the mIgG or supCD28 mAb group recipients. The supCD28 mAb-expanded splenocytes transferred recipient showed no evidence of the GvHD (right), whereas the marked lymphocytes infiltrations and tissue injury were seen in the control Lewis splenocytes transferred (left). Original magnification 100×. (B) FCM analysis showed a progressive increased of the CD4+CD25+ T-cell population on day 14 after transfer of control splenocytes transferred, and a decrease in Foxp3+ cells decreased. The population of the Foxp3+ cells in CD4+CD25+ T cells and total T cells showed to be maintained at a higher level.
In Vivo supCD28 mAb-Expanded T-Cell Suppressed Systemic GvH Reactions *

Systemic GvH reaction were performed from Lewis T cells to F1 (Lewis × DA) rats.
p-Value was compared with naive group rat and determined by the Kaplan-Meier test.
High Level of Foxp3-Expressing Cells in Peripheral Blood (PB) After Transfer supCD28 mAb-treated Lymphocytes
A kinetic analysis of Foxp3+ cells in the peripheral blood of the GvHD host was conducted. The FCM analysis showed a progressive increase of the CD4+CD25+ population on day 14 after transfer of control splenocytes (Fig. 5B, upper) and a decrease in the number of Foxp3+ cells, thus indicating an expansion of CD25+ FoxP3- effector cells in response to the alloantigens encountered (Fig. 5B, middle). In contrast, the population of the CD4+CD25+ T cells of the host peripheral blood in the group of supCD28 mAb-expanded splenocytes only, or mixed at an equal ratio of naive Lewis splenocytes transferred showed no change until 56 days (Fig. 5B, lower). Furthermore, the population of the Foxp3+ cells in CD4+CD25+ T cells and total T cells were maintained at a higher level (Fig. 5B, middle and lower). These data indicate that in the presence of the transferred supCD28 mAb-expanded CD4+CD25+Foxp3+ cells, the response of effector cells resulting in lethal GvHD is inhibited.
Discussion
The ultimate goal of organ transplantation is the modulation of the immune response to produce tolerance without immunosuppressive agents, and accumulating evidence suggests that Treg cells play an important role in inducing and maintaining transplantation tolerance (19,22). Therefore, different strategies have been sought to induce tolerance and prevent acute and chronic rejection. The present study documents, for the first time, that supCD28 mAb could expand Foxp3-expressing Treg cells to suppress rat GvH reactions and inhibit cardiac allograft rejection.
Consistent with previous reports (21,26), the current data showed that supCD28 mAb stimulates all resting rat T cells to proliferate in vitro and in vivo in the absence of TCR ligation and that supCD28-expanded T cells express higher levels of IL-2, –4, and –10 and lower levels of INF-γ mRNA (Fig. 3). In addition, supCD28 mAb treatment markedly decreased the transcript level of perforin and granzyme B, which are well-established markers of CTL activity and allograft rejection (data not shown). Consistent with a whole expanded T cell analysis, unlike conventionally activated CD4+CD25- T cells that express IL-2 and IFN-γ, isolated supCD28 mAb-expanded CD4+CD25+ T cells produced less IFN-γ and instead preferentially produced IL-4 and IL-10 (Fig. 3) as described previously (16). Expanded CD4+CD25+ T cells not only suppressed antibody-induced lymphocyte proliferation in agreement with Lin's report (16), but also in agreement with the report by Beyersdorf et al., which inhibited the response to ConA (4) and to MHC-disparate stimulator cells in vitro (Fig. 2).
The PLN assay, a well-accepted assay for local GvH activity and a systemic GvHD model revealed that, consistent with the in vitro results, supCD28 mAb-expanded CD4+CD25+ T cells had marked suppressive activity against co-injected naive lymphocytes. Furthermore, they exhibited regulatory efficacy against co-injected naive lymphocytes, resulting in suppression of GvHD (Fig. 4). The inhibitory effect of expanded CD4+CD25+ T cells against inflammatory responses correlated with the persistence of a high level of Foxp3-expressing T cells in the host peripheral blood (Fig. 5). As described previously (13), nTreg cells specifically express the transcription factor Foxp3, which appears to act as a master control gene for their development and function. Forced expression of the Foxp3 gene can convert mouse naive T cells to Treg cells that phenotypically and functionally resemble nTreg cells (13). Taken together, these data suggest that the capacity of the in vivo expanded CD4+CD25+ T cells to suppress GvH activity was correlated with Foxp3 expression.
In addition to being a marker for Treg cells, CD25 expression on CD4+ T cells is an indicator of cell activation. Specifically, stimulation of CD4+CD25- T cells leads to several outcomes, including proliferation, cytokine production, and induction of cell surface expression of CD25. It is, therefore, possible that some or all of the CD4+CD25+ T cells are the result of recent activation. To investigate the origins and the relative expression of Foxp3, expressed by the expanded Treg cells after supCD28 mAb stimulation, an assay was performed to determine whether CD4+CD25- T cells that expressed CD25 after activation in vitro showed Foxp3 expression. Therefore, CD4+CD25- T cells were activated with plate-bound supCD28 mAb and after 72 h the population of CD4+CD25+ activated T cells were detected. After antibody stimulation, all activated cells expressed CD25, whereas Foxp3 expression was not found (Fig. 3C). Furthermore, consistent with Beyersdorf et al. (4), all in vitro supCD28-expanded rat CD4+CD25+ Treg cells remained positive for not only CD25 but also for high Foxp3 expression, while expression of FoxP3 was induced in very few CD4+CD25+ T cells derived from CD25- T cells. Moreover, there was no significant difference in the proliferative response to antibody stimulation between the activated Foxp3+ or Foxp3- T cells (data not shown). Together with the data of Lin and Hünig (16), who failed to detect acquisition of CD25 on in vivo-stimulated CD4+CD25- cells and hence concluded a preferential expansion of preexisting nTreg cells, the current data clearly indicate that supCD28 mAb has the unique capacity to expand intrinsic constitutively CD25- and Foxp3-expressing nTreg cells, which are not activated CD25+ effector T cells.
In vivo-expanded CD4+CD25+ T cells with suppressive capacity were derived from preexisting nTreg cells constitutively expressing CD25 and Foxp3 in vivo, in contrast to results seen with in vitro stimulation where T cells became CD25+ after in vitro activation in response to supCD28 but did not express Foxp3. These data, which be correlated with Lin and Hünig's (16), indicate that, unlike what has been described in mice for antigen-driven conversion of CD4+25- to Treg cells (15), stimulation of CD4+CD25- T cells through the supCD28 mAb stimulation results in the expansion of preexisting CD4+CD25+ Treg cells.
IL-2 is a cytokine indispensable for the survival of nTreg cells in the peripheral immune system; and CD25 is also indispensable, as it is a constituent of the high-affinity IL-2 receptor (18). Therefore, IL-2 signaling seems to be critically required for maintaining the homeostasis and competitive fitness of Treg cells in vivo. The present study demonstrated that IL-2 gene transcription was upregulated and hence IL-2 production was increased after supCD28 stimulation in vivo (Fig. 3). Recent studies have concluded that the functional significance of CD25 expression on Treg cells is a requirement for IL-2 in their generation or survival (24,27). Furthermore, in vitro studies have shown that Treg cells rapidly lose viability in the absence of IL-2. Therefore, the present data suggest that IL-2 may play an important role in supCD28 mAb-mediated Treg expansion in vivo.
In conclusion, the current results confirm that supCD28 mAb expands rat Foxp3 expressing CD4+CD25+ nTreg cells in vivo, which maintain their unique cell surface marker profile and inhibit alloreactive T-cell proliferation. Moreover, these expanded CD4+CD25+ Treg cells potently inhibit the GvH activity. These results, therefore, illustrate again and in a new setting the therapeutic power of transient polyclonal activation of Treg cells in vivo. While the recent attempt to translate supCD28-mediated nTreg stimulation failed because of the unexpected induction of a life-threatening cytokine release syndrome (25), transient polyclonal Treg activation, regardless of the tool used for its induction, still represents an attractive therapeutic option in autoimmunity and transplantation tolerance. Regardless of the feasibility of direct in vivo expansion, the ability to isolate and expand human CD25+CD4+ nTreg cells in vitro for reinfusion will allow the further biological and biochemical characterization of this unique T-cell subset and expedite progress towards the clinical use of Treg cells as a cellular therapy for the treatment of allograft rejection and GvHD.
Footnotes
Acknowledgments
The authors thank Naoko Funeshima-fuji for her scientific discussion and technical assistance. This study was supported by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (a Grant-in-Aid 17390355, 20390349), and Ministry of Health, Labour and Welfare of Japan (HS KHC1025), and Wilhelm-Sanderstiftung and Bavarian Research Foundation.