
Abstract
Introduction
Diosbulbin B (DB) has emerged as a potential drug for tumor treatment, but its hepatotoxicity restricts clinical use. The role of autophagy in DB-induced liver damage remains unclear. This study investigates autophagy mechanisms in DB hepatotoxicity and the protective effect of Rosa roxburghii Tratt juice (RRTJ).
Methods
Rats were randomized into control group, DB (12.5, 25, 50 mg/kg), RRTJ control, and RRTJ + DB (5/10 mL/kg + 50 mg/kg), and orally administered for 1 month. Hepatic superoxide dismutase (SOD)/glutathione peroxidase (GSH-Px) activities, malondialdehyde (MDA) content, serum liver function markers, and liver histopathology were assessed. Gene and protein expression levels of Beclin1, LC3B, and p62 were detected by real-time quantitative polymerase chain reaction (qPCR), immunohistochemistry, and Western blot. QPCR and immunohistochemistry evaluated the potential role of AMPK.
Results
25 and 50 mg/kg DB induced liver damage by reducing the levels of SOD/GSH-Px in liver tissues and increasing MDA. Resulting in elevated serum AST levels and histopathological changes such as cytoplasmic loosening, punctate necrosis in hepatocytes. DB-induced liver injury was associated with autophagic dysfunction. Compared with the control group, DB-treated rats showed upregulated expression of autophagy-related proteins Beclin1, LC3B, and p62. RRTJ inhibited DB-induced AMPK phosphorylation and reduced the expression of Beclin1, LC3B, and p62, thereby attenuating liver injury.
Conclusion
DB induces liver injury via autophagy dysregulation, while RRTJ alleviates damage by reducing AMPK phosphorylation to modulate autophagy, suggesting RRTJ as a potential strategy against DB-induced hepatotoxicity.
Introduction
Dioscorea bulbifera L. (DBL), a traditional Chinese medicinal herb, is recognized for its anti-inflammatory, antioxidant, and immune-regulatory properties. One of its primary active components, diosbulbin B (DB), is a diterpene lactone with a furan ring structure.1,2 DB has demonstrated significant antitumor potential in preclinical studies.3–5 However, prior research indicates that low doses of DB exhibit limited antitumor efficacy, whereas high doses induce severe hepatotoxicity, thereby restricting its clinical translation.6,7 Therefore, elucidating the mechanisms of DB-induced liver injury and identifying protective agents are critical for advancing its clinical application.
Autophagy, a highly conserved cellular degradation process, is essential for maintaining hepatic homeostasis. Autophagosomes, which are double-membrane structures, encapsulate aged or damaged organelles in response to stimuli such as hypoxia or starvation. These autophagosomes then fuse with lysosomes to form autolysosomes, where the contents are degraded, thereby maintaining cellular stability and promoting organelle renewal.8,9 In vitro studies have demonstrated that DB induces reactive oxygen species (ROS) accumulation and triggers autophagy via mitochondrial damage. 7 Conversely, conflicting evidence from primary hepatocyte models suggests that autophagy may not be activated during DB-induced injury, 10 highlighting the need to clarify the role of autophagy in DB-mediated liver damage.
AMP-activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine kinase that maintains cellular energy homeostasis under low-energy conditions by regulating anabolic and catabolic pathways. 11 AMPK is activated in response to mitochondrial damage and participates in mitochondrial quality control by modulating mitophagy. 12 Although previous studies have shown that DB impairs mitochondrial function by inhibiting Na+-K+-ATPase, Ca2+-Mg2+-ATPase, and total ATPase activities—thereby inducing mitochondrial sodium-water retention, calcium overload, and magnesium depletion 13 —the interplay between mitochondrial dysfunction and AMPK activation in the context of DB-induced liver injury remains undefined and requires further mechanistic exploration.
Increasing evidence suggests that natural products offer a significant advantage in preventing and treating liver injury. 14 Rosa Roxburghii Tratt (RRT), a unique wild fruit native to southwestern China, is rich in nutrients such as superoxide dismutase, flavonoids, polyphenols, and triterpenoids. 15 Studies have demonstrated that RRTJ can alleviate non-alcoholic fatty liver disease by enhancing mitochondrial function. 16 Additionally, RRTJ has been shown to enhance antioxidant defenses and mitigate arsenic-induced liver damage.17,18 These original findings indicate that RRTJ may serve as a potential antidote for DB-induced liver injury.
To explore this potential, a rat model of DB-induced liver injury was established, assessing the levels of antioxidant enzymes (SOD, GSH-Px) and the lipid peroxidation product MDA in liver tissue. Transcriptional and protein expression levels of AMPK and autophagy-related proteins (Beclin1, LC3B, p62) were quantified, alongside liver function biomarkers (ALT, AST) and histopathological analyses, to elucidate the role of the AMPK-autophagy axis in DB-mediated hepatotoxicity. Additionally, RRTJ was administered to DB-exposed rats to evaluate its protective effects and underlying mechanisms via parallel analyses of the aforementioned parameters. This study provides experimental evidence for the development of novel therapeutic strategies against DB-induced liver injury.
Materials and methods
Drug preparation
Diosbulbin B (PCS0503, ≥90% purity) was purchased from Chengdu Phytochemical Pure Biotechnology Co., Ltd (China). As described in the literature, 6 DB was suspended with 0.5% sodium carboxymethyl cellulose (CMC–Na) (final volume: 50 mL) and stored at 4°C until use. RRTJ was obtained from China National Pharmaceutical Group Guizhou Health Industry Development Co., Ltd (health food license: National Health Commission of China [2002]0004).
Experimental animals
Eight-week-old healthy specific pathogen-free (SPF) Sprague-Dawley (SD) rats (200 ± 20 g) were purchased from Changsha Tianqin Biotechnology Co., Ltd (China; license number: SCXK (Xiang) 2019-0014). The animals were housed in a controlled environment with a temperature of 22–24°C, relative humidity of 40–60%, and a 12/12 h light/dark cycle, with free access to standard rodent chow and tap water. All animal procedures were approved by the Laboratory Animal Ethics Committee of Guizhou Medical University (Approval No.: 2303062) and performed in accordance with the Guide for the Care and Use of Laboratory Animals.
The rats were randomly divided into groups as follows: control group (0.5% CMC-Na); DB (12.5, 25, 50 mg/kg) groups; intervention group of RRTJ and control group of RRTJ. The intervention groups of RRTJ were the low-dose RRTJ group (5 mL/kg) + 50 mg/kg DB and the high-dose RRTJ group (10 mL/kg) + 50 mg/kg DB. The selection of the exposure dose and exposure time of DB was designed based on the literature19,20; the choice of the dose of RRTJ was based on the literature,21,22 as well as the previous studies of our research group. 17 Each group consisted of 10 rats, with an equal distribution of males and females. All rats were administered orally for 1 month. Rats were monitored daily for signs of distress, and all efforts were made to minimize suffering. Pentobarbital sodium (0.2 mg/kg, i.p.) was used for terminal anesthesia, and rat blood and liver tissue were collected.
Serum alanine aminotransferase/aspartate aminotransferase (ALT/AST) activity analysis
Serum was obtained from blood samples following cryogenic centrifugation at 3000 rpm for 15 min. The activities of ALT and AST in the serum were measured using a fully automated serum analyzer.
The level of serum glucose
Serum was obtained from blood samples following cryogenic centrifugation at 3000 rpm for 15 min. The glucose concentrations in the serum were measured using a fully automated serum analyzer.
Histological evaluation of the liver
Liver tissues from the same part were fixed in 4% paraformaldehyde at 4°C overnight. After dehydration, they were embedded in paraffin to prepare conventional sections for hematoxylin and eosin (H&E) staining. Finally, liver tissue histopathological changes were performed and recorded under an optical microscope.
The activity assays of liver tissues of SOD, MDA, GSH-Px
The liver tissues were diluted with ice-cold physiological saline and homogenized with a homogenizer to obtain homogenates. The contents of SOD (A001-3), MDA (A003-1), and GSH-Px (A005-1) in the liver tissues were determined using kits from Nanjing Jiancheng Bioengineering Institute under the manufacturer’s instructions.
Immunohistochemical analysis
After deparaffinization with xylene and rehydration with a gradient alcohol, the paraffin sections of 4 μm were boiled in citrate buffer for antigen retrieval. When the slices were cooled to room temperature, 3% hydrogen peroxide was applied to quench endogenous peroxidase in the dark. The sections were incubated with the corresponding primary antibodies Beclin1 (Abcam, ab62557, 1:250), LC3B (Abcam, ab192890, 1:200), p62 (Abcam, ab91526, 1:200), p-AMPK (CST, 2535, 1:200) at 4°C overnight in a humid cassette after blocking with 10% goat serum at room temperature for 30 min. Sections were incubated with goat anti-rabbit IgG (ab205718, 1:2000) at 37°C for 30 min. Finally, diaminobenzidine (DAB, ZSGB-BIO, ZLI-9017) chromogen was added before counterstaining with hematoxylin at room temperature for 3 min. Then, samples were analyzed under a light microscope. The intensity of positive areas from 10 random fields (magnification, 200x) in each section was quantified using Image-Pro Plus 6.0 analysis software.
Transmission electron microscopy
The rat liver tissue was pre-fixed in 3% glutaraldehyde for 48 h and subsequently fixed in 1% osmium tetroxide for 1 h. Subsequently, it was dehydrated with acetone step by step and finally embedded in Epon 812. The semithin sections were stained with methylene blue. The ultrathin sections were cut with a diamond knife and stained with uranyl acetate and lead citrate. The sections were examined using a JEM-1400-FLASH transmission electron microscope.
Western blot analysis
As previously described, 23 total protein was extracted from liver tissues using RIPA lysis buffer, and the protein concentration was determined through a BCA protein quantification kit. Subsequently, protein electrophoresis was conducted using a 12% SDS-PAGE gel. The separated proteins were then transferred onto a PVDF membrane, which was incubated at 4°C overnight with primary antibodies against Beclin1 (1:2000), LC3B (1:1000), p62 (1:1000), and β-actin (1:4000). The PVDF membrane was exposed to a secondary antibody for 2 h at room temperature. Chemiluminescent signals were generated using an ECL solution and captured with the Bio-Rad ChemiDoc imaging system (USA). Quantify the band intensity using Image Lab 6.0 software, and normalize the expression of the target protein to that of β-actin from the same sample.
Real-time PCR analysis
Total RNA was extracted from liver tissues using TRIzol reagent (Invitrogen, USA) following the manufacturer’s protocol. Then, cDNA was obtained using the PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, RR047A) according to the manufacturer’s instructions. Finally, the Bio-Rad CFX96 machine (Bio-Rad, USA) detected Beclin1, LC3B, p62, and AMPK genes according to cyclic thresholds. Relative gene expression was normalized to GAPDH and calculated using the 2−ΔΔCq method. 24 The Shanghai Institute of Biological Engineering (China) designed and synthesized the primers. Table (S1.rtf, Table 1) lists the primers used in real-time qPCR.
Statistical analysis
All data were analyzed using Excel 2019 and SPSS Statistics 25.0. Normality of data distribution was assessed using the Shapiro-Wilk test, and homogeneity of variances was evaluated via Levene’s test. For datasets satisfying normal distribution and homoscedasticity assumptions, one-way analysis of variance (ANOVA) was performed to compare differences across groups. Following significant ANOVA results, the least significant difference (LSD) post-hoc tests were conducted. Data are presented as mean ± standard deviation (SD), and statistical significance was defined as p < 0.05 after correction.
Results
RRTJ decreased the DB-induced increase in serum levels of ALT, AST
As illustrated in Figure 1, male rats treated with DB for 1 month showed a dose-dependent elevation in serum ALT levels compared to controls, though these changes did not achieve statistical significance (p > 0.05). In contrast, serum AST levels in male rats treated with 25 and 50 mg/kg DB showed significant increases (p < 0.05). Co-administration of RRTJ (5–10 mL/kg) effectively attenuated increased serum ALT and AST levels (p < 0.05). Previous studies have demonstrated sex-specific differences in DB hepatotoxicity,
25
consistent with our finding that female rats showed no significant AST/ALT elevation at any DB dose (S1.rtf Figure 1). This is likely due to lower hepatic CYP3A1/2 activity in females.
26
These results highlight the importance of using male rats to model CYP3A1/2-dependent DB toxicity, as females may require higher doses or longer exposure to induce comparable liver injury. Considering that the expression of CYP3A1/2 in female rats may be lower,
26
and the sample size of female rats in this study was relatively small, to focus on the more definite dose-response relationship in male rats, only the data of male rats were included in the subsequent analysis. RRTJ alleviates liver injury induced by DB in rats. (a) ALT levels; (b) AST levels. n = 5 for all groups except Group 2 (n = 4, one sample lost during processing). Missing data in Group 2 were replaced with the group mean for statistical consistency. The statistical analysis was done using one-way ANOVA and the LSD test. *p < 0.05 versus control group; #p < 0.05, ##p < 0.01 versus DB 50 mg/kg group. Data are presented as mean ± SD.
Histological evaluation of liver
Histological analysis was performed to evaluate the histopathological effects of DB on rat liver and the protective efficacy of RRTJ. As shown in Figure 2, hepatocytes in the control group displayed an orderly arrangement, distinct nuclei, and no apparent pathological changes. Treatment with 12.5 mg/kg DB for 1 month induced no significant histopathological alterations. In the 25 mg/kg DB group, inflammatory cell infiltration was observed in the portal area, accompanied by hepatocyte cytoplasmic laxity and focal nuclear pyknosis. The 50 mg/kg DB group exhibited marked hepatocyte cytoplasmic loosening and numerous conspicuously pyknotic liver cell nuclei in the periportal region. These findings demonstrate that 25 and 50 mg/kg DB caused hepatic injury in rats after 1 month. Conversely, co-administration of 5 mL/kg and 10 mL/kg RRTJ with 50 mg/kg DB significantly alleviated the histopathological damage in rat liver tissues. The RRTJ monotherapy group showed no significant pathological changes, suggesting that RRTJ mitigates DB-induced liver injury. RRTJ improves pathological injury of liver tissues induced by DB in rats. n = 5 for all groups except Group 2 (n = 4, one sample lost during processing). Scale bar = 50 μm. The cyan arrows indicate inflammatory cells, the dark gray arrows indicate hepatocellular necrosis, and the blue arrows indicate hepatocellular edema.
RRTJ reversed DB-changed expression of SOD, GSH-Px, MDA
Oxidative stress induced by DB was evaluated by measuring MDA, SOD, and GSH-Px levels in liver tissues of each treatment group using commercial assay kits. Compared with the control group, the 50 mg/kg DB group showed significantly higher hepatic MDA levels (p < 0.01). Treatment with 25 mg/kg and 50 mg/kg DB significantly reduced SOD and GSH-Px activities (p < 0.05), indicating that 1 month administration of these DB doses induced oxidative damage in rats. Following RRTJ intervention, 5 mL/kg RRTJ tended to decrease hepatic MDA content and increase SOD/GSH-Px levels in the 50 mg/kg DB-treated group, although these changes were not significant (p > 0.05). In contrast, 10 mL/kg RRTJ significantly enhanced SOD and GSH-Px activities while reducing MDA levels (p < 0.05 for SOD/GSH-Px; p < 0.01 for MDA), suggesting that 10 mL/kg RRTJ alleviates DB-induced liver injury via antioxidant effects (Figure 3). RRTJ alleviates oxidative damage induced by DB in rats through antioxidation. (a) MDA levels; (b) GSH-Px activity; (c) SOD activity. n = 5 for all groups except Group 2 (n = 4, one sample lost during processing). Missing data in Group 2 were replaced with the group mean for statistical consistency. The statistical analysis was done using one-way ANOVA and the LSD test. *p < 0.05 versus control group; #p < 0.05, ##p < 0.01 versus DB 50 mg/kg group. Data are presented as mean ± SD.
Effects of DB on the microstructure of liver tissue
The results demonstrate that 1 month exposure to DB induces oxidative damage in rat liver tissue. To investigate the role of autophagy in DB-induced liver injury, the ultrastructure of liver tissues was analyzed using transmission electron microscopy (TEM). In the control group, hepatocytes exhibited normal morphology and structure, with only occasional lysosomes observed. In rats treated with 12.5 mg/kg DB, slight lysosomal accumulation was noted. Conversely, 25 mg/kg and 50 mg/kg DB treatment caused significant mitochondrial condensation, endoplasmic reticulum dilation, and prominent formation of lysosomes and autophagosomes in hepatocytes (Figure 4). These observations indicate that DB induces mitochondrial damage and triggers autophagic responses in rat liver. DB-induced mitochondrial damage and autophagic activation in hepatocytes (a) Control; (b) 12.5 mg/kg; (c) 25 mg/kg; (d) 50 mg/kg. Transmission electron microscopy images show ultrastructural changes in DB-treated hepatocytes (n = 3). Nucleus (N), mitochondria (Mi), rough endoplasmic reticulum (RER); The green arrow indicates hepatocytes with mitochondrial shrinkage, the blue arrow indicates chromatin lysis, the black arrow indicates lysosomes, and the red arrow indicates autophagy (scale = 1.0 μm).
The RRTJ alleviated the autophagy impairment of liver tissue in rats caused by DB
The TEM observations demonstrated that DB treatment triggered autophagic responses in rat liver. To further characterize the effect of DB on autophagy, the expression levels of autophagy-related proteins (Beclin1, LC3B, and p62) were analyzed by immunohistochemistry. As shown in Figure 5(a), Beclin1, LC3B, and p62 were predominantly localized in the cytoplasm. Compared with the control group, 25 mg/kg and 50 mg/kg DB treatment induced Beclin1 activation and significantly increased the autophagic marker LC3B levels (p < 0.05 and p < 0.01, respectively). Conversely, the autophagy substrate p62 did not decrease; instead, it showed a significant increase in DB-treated groups compared to the control (p < 0.05) (Figure 5(b)), suggesting impaired autophagic flux. Notably, administration of 5 mL/kg and 10 mL/kg RRTJ significantly reduced the expression of Beclin1, LC3B, and p62 in the 50 mg/kg DB-treated group (p < 0.05), indicating that RRTJ alleviates DB-induced autophagic dysfunction. The effects of RRTJ treatment on autophagic protein expression in rats treated with DB. (a) Immunohistochemistry for Beclin1, LC3B, and p62 in liver tissue (magnification, 200x). Scale bar = 50 μm. (b) Photo densitometry was performed on picture A by immunohistochemistry. n = 5 for all groups except Group 2 (n = 4, one sample lost during processing). Missing data in Group 2 were replaced with the group mean for statistical consistency. The statistical analysis was done using one-way ANOVA and the LSD test. *p < 0.05, **p < 0.01, ***p < 0.01 versus control group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus DB 50 mg/kg group. Data are presented as mean ± SD.
Western blot analysis showed that Beclin1 expression was upregulated in liver tissues of rats treated with 25 mg/kg and 50 mg/kg DB compared to the control group. Additionally, the LC3B II/I ratio was significantly increased in the 50 mg/kg DB group; however, the autophagy substrate p62 did not exhibit significant degradation (p < 0.05). qRT-PCR results revealed that 50 mg/kg DB significantly induced Beclin1 and LC3B mRNA expression (p < 0.05) while not affecting p62 mRNA levels (p > 0.05). Conversely, treatment with 5 mL/kg and 10 mL/kg RRTJ suppressed Beclin1 and p62 protein expression and inhibited the conversion of LC3B I to LC3B II. qPCR data corroborated these findings, showing no significant changes in p62 gene expression (p > 0.05). Collectively, these results suggest that RRTJ alleviates DB-induced liver injury in rats by regulating autophagic flux (Figure 6(a)–(g)). The effects of RRTJ treatment on autophagic protein and gene expressions in DB-treated rats. (a)The expression of Beclin1, LC3B, and p62 proteins. (b–d) Densitometric quantification of protein bands shown in Figure A. n = 3 for Western blot, all results were obtained from at least 3 independent experiments. (e–g) The mRNA levels of Beclin1, LC3B, and p62. (n = 4 for qPCR; relative expression normalized to GAPDH and expressed as fold change vs. control.). *p < 0.05, **p < 0.01 versus control group; #p < 0.05, ##p < 0.01, versus DB 50 mg/kg group. Data are presented as mean ± SD.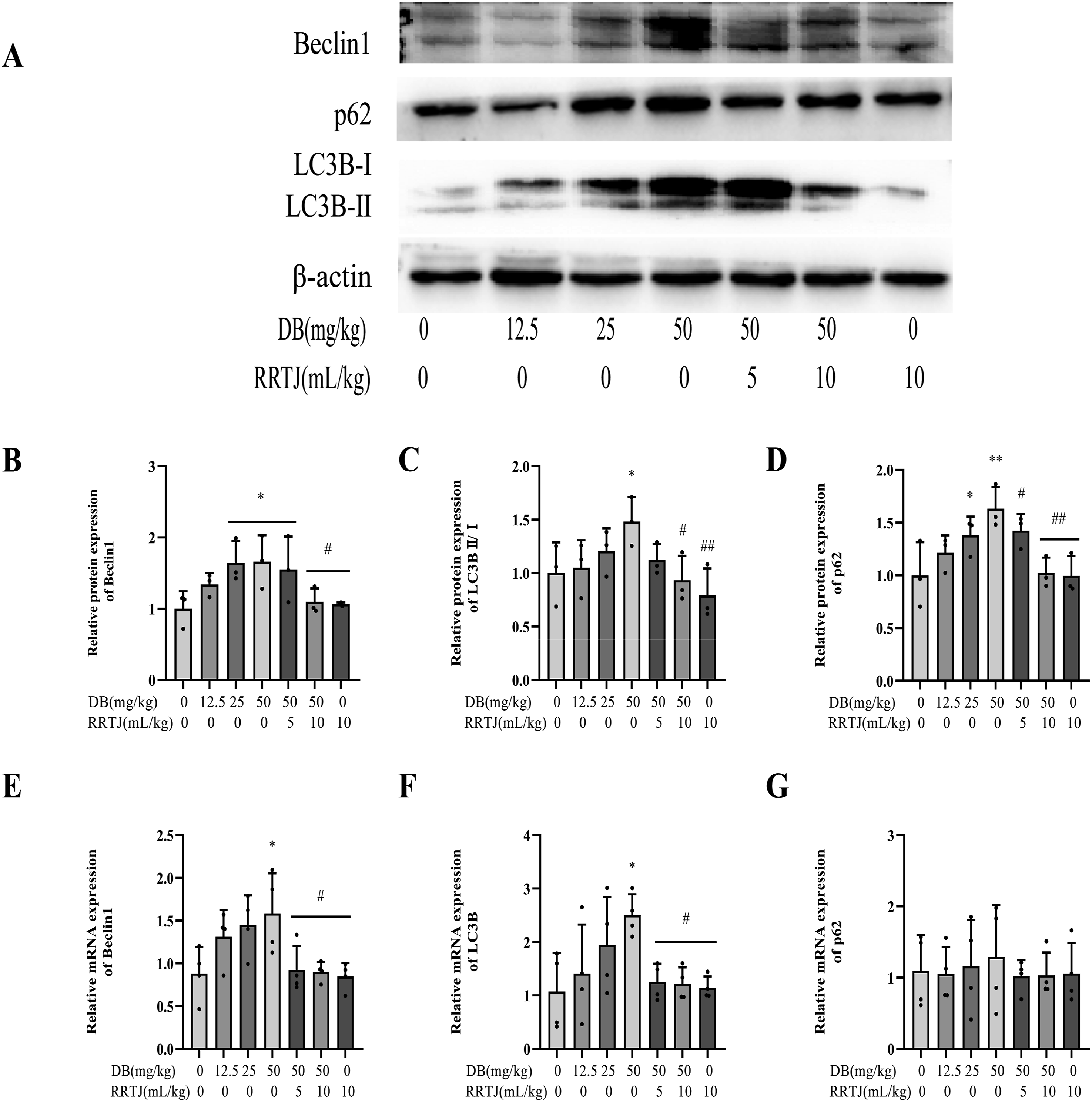
Influence of RRTJ on AMPK induced by DB
AMPK plays a critical role in regulating energy metabolism and autophagy. To investigate the potential mechanism of AMPK involvement in DB-induced liver injury, its gene and protein expression levels were analyzed by qPCR and immunohistochemistry. As depicted in Figure 7, the phosphorylated AMPK (p-AMPK) protein was predominantly expressed in the cytoplasm. Treatment with DB at doses of 25 mg/kg and 50 mg/kg significantly increased the phosphorylation of the AMPK protein (p < 0.01). Additionally, qPCR results showed upregulated expression of the AMPK gene, indicating that DB-induced liver injury may activate the AMPK signaling pathway. Conversely, the expression of AMPK mRNA and its protein phosphorylation could be reversed with the intervention of 5 mL/kg and 10 mL/kg RRTJ (p < 0.05, p < 0.01). RRTJ downregulates AMPK signaling in the liver of DB-induced rats. (a) The expression of p-AMPK proteins (magnification, 200x). Scale =50 μm; (b) Photo densitometry was performed on picture A by immunohistochemistry (n = 4); (c) The mRNA levels of AMPK. The relative expression levels were normalized to the housekeeping gene GAPDH and presented as fold-change compared to the control group (n = 4). The statistical analysis was done using one-way ANOVA and the LSD test. *p < 0.05, **p < 0.01 versus control group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus DB 50 mg/kg group. Data are presented as mean ± SD.
Discussion
This study aims to investigate the role of autophagy in liver injury induced by DB and to explore the intervention effects of RRTJ in vivo. The results reveal that treatment with DB results in oxidative damage, mitochondrial impairment, and autophagy dysfunction within rat models. RRTJ administration exerted antioxidant effects, restored autophagy flux, and attenuated DB-induced liver injury.
Serum AST and ALT levels are sensitive indicators for assessing liver function.27,28 Previous studies have reported gender differences in DB-induced hepatotoxicity. 25 In this study, male rats were more sensitive to DB-induced hepatotoxicity: 50 mg/kg DB significantly increased serum AST levels in males, whereas ALT levels remained unchanged. Serum AST elevation reflects mitochondrial injury—a critical marker of severe hepatic damage, 29 which is related to mitochondria being the target cells of DB. The serum levels of AST in male rats exhibited more pronounced alterations, which may be associated with sex differences in hepatic CYP450 enzyme activity. CYP3A subfamily enzymes (e.g., CYP3A1/2) are 2–3-fold more active in male rats than in females, 26 mediating DB metabolic activation to generate electrophilic toxic intermediates that induce hepatocellular injury. Females may be protected by estrogen-regulated pathways: Estrogen inhibits CYP3A4 transcription via ERRα receptor activation, 30 and alleviates mitochondrial dysfunction induced by mitochondrial oxidative stress. 31 Female rats showed elevated hepatic GSH levels, enhanced GCL/GPx activity, and reduced serum ALT/AST compared to males. 32 Progesterone fluctuations during the estrous cycle could modify hepatic P450 profiles, explaining the inter-individual variability in DB metabolism (e.g., the higher standard deviation in female rats treated with DB 12.5 mg/kg). In this study, female rats did not exhibit significant AST/ALT elevation at any DB dose. Sex-stratified analysis did not reach statistical significance (p > 0.05), though this may reflect the limited sample size rather than a true biological equivalence. Histological analysis showed that 25 and 50 mg/kg DB could lead to necrosis of rat hepatocytes with edema, loose cytoplasm, and nuclear contraction, consistent with related studies.33,34
Oxidative stress is recognized as one of the primary mechanisms responsible for liver injury induced by DB. In vivo, DB is metabolically activated by CYP3A4, generating a reactive intermediate that covalently binds to proteins, nucleic acids, and undergoes electrophilic conjugation with glutathione (GSH), ultimately leading to liver toxicity.35–37 GSH depletion triggers excessive ROS accumulation, resulting in lipid peroxidation (e.g., elevated MDA).38,39 Antioxidant enzymes (e.g., SOD and GSH-Px) play a pivotal role in scavenging intracellular free radicals, minimizing ROS formation, and mitigating lipid peroxidation to preserve cell membrane integrity. 40 For the assessment of liver injury, the activities of MDA, SOD, and GSH-Px were determined. Results showed that 50 mg/kg DB significantly increased hepatic MDA levels while decreasing SOD and GSH-Px activities, indicating that oxidative stress contributes to DB-induced liver injury in rats.
Autophagy is an essential cellular protective mechanism through which excess or damaged organelles are degraded.41,42 Oxidative-antioxidant imbalance generates excessive reactive oxygen species (ROS; e.g., superoxide anions, H2O2), which damage macromolecules and organelles. 43 In response, autophagy is activated to eliminate these dysfunctional organelles. 44 DB treatment induced ultrastructural pathologies in hepatocytes, including mitochondrial cristae disruption, chromatin margination, and autophagosome-lysosome proliferation, consistent with previous studies. 7 Beclin1 serves as a scaffold for the phosphatidylinositol 3-kinase (PI3K) complex, marking the initiation of autophagosome formation. 45 The conversion from LC3BI to LC3BII reflects the overall level of autophagosome biogenesis, while p62 (SQSTM1), autophagic substrate adaptor protein, is closely associated with autophagic flux efficiency. Under normal conditions, lysosomes degrade p62 during autophagy. However, when the fusion of autophagosomes and lysosomes is impaired, p62 accumulates. 10 Notably, 25 and 50 mg/kg DB treatment significantly upregulated the protein expression of Beclin1, LC3B, and p62. The increase in LC3BII serves as an indicator of activated autophagosome biogenesis, which represents the cell’s initial stress response to DB toxicity. However, p62—a protein normally degraded during autophagic flux—exhibited concurrent and persistent overexpression. This directly indicates autophagic malfunction. Collectively, co-upregulation of LC3B and p62 is a crucial biomarker for autophagic flux blockade, rather than mere autophagy activation. This paradox is explained by the following: Upon DB exposure, autophagy is initially activated as a self-defense mechanism to mitigate cellular stress. Concurrently, DB disrupts downstream autophagy, likely by interfering with autophagosome-lysosome fusion or lysosomal function. Consequently, although autophagosomes are actively formed, as evidenced by the elevated levels of LC3BII, the degradation process remains incomplete. Inability to degrade autophagic cargo leads to p62 accumulation, a hallmark of impaired autophagic flux. 46 This duality arises because DB triggers autophagy as a stress response while disrupting downstream flux, such as autophagosome-lysosome fusion or lysosomal function. Concurrently, DB may disrupt downstream autophagic processes, such as autophagosome-lysosome fusion or lysosomal function. Thus, while autophagosomes form, they cannot complete degradation, resulting in the accumulation of autophagic substrates like p62. p62 accumulation impairs proteasomal function, causing re-recruitment of ubiquitinated substrates into autophagy. 47 This exacerbates lysosomal degradation workload. Impaired autophagy causes cellular damage/death and promotes liver disease progression, validated in NAFLD patient livers, hepatocytes, endothelial cells, and hepatic stellate cells.48–50 Dysregulated autophagy is implicated in diverse diseases, including cancer and neuropathies. 51 During nervous system injury, disruptions in lysosomal acidification and Ca2+ regulation impair autophagy, causing lysosomal storage disorders. 52 These impairments ultimately lead to neuronal cell necrosis. 53 Impaired autophagy prevents timely clearance of amyloid or aggregation-prone proteins, accelerating Alzheimer’s disease (AD) progression. 54 During defective autophagy, both protein aggregates and their autophagic cargo receptor p62 (normally degraded via autophagy) accumulate. This triggers cellular stress responses—including ROS production, ER stress, and DNA damage activation—ultimately promoting tumorigenesis.55–58
Adenosine monophosphate-activated protein kinase (AMPK) regulates body metabolism, allowing cells to respond to internal and external environmental alterations (such as ischemia, nutrient deprivation, hypoxia, and oxidative stress) via phosphorylation.59–61 Upon mitochondrial damage, AMPK is activated by sensing reduced ATP production. 62 It regulates mitochondrial fission by directly phosphorylating mitochondrial fission factor (MFF) and activating autophagy.63,64 Studies show that DB treatment elevates ROS levels, decreases membrane permeability, and impairs cellular bioenergetics. 7 Studies show that ROS activates AMPK by lowering glucose levels, promoting autophagic energy recycling. 65 Treatment with DB significantly reduced serum glucose levels in rats (S1.rtf Figure 2). Additionally, rats treated with 50 mg/kg DB showed significantly increased hepatic AMPK gene expression, and immunohistochemistry revealed elevated p-AMPK levels, indicating DB activates AMPK under glucose deprivation. Studies have shown that ROS can activate AMPK by reducing glucose levels to drive the autophagic recycling of energy resources. 66 While AMPK activation is linked to lysosomal acidification impairment in glucose-deprived cells, 65 AMPK knockdown had minimal effect on this impairment, 67 suggesting a complex role for AMPK in autophagic flux regulation.
RRT is a distinctive natural medicinal and edible plant in the southwest region of China, rich in bioactive components including vitamins, minerals, flavonoids, and Kaji-ichigoside F1.
68
Previous research has shown that RRTJ can mitigate arsenic-induced liver injury by regulating elemental balance and oxidative stress.17,18 Given the lack of effective therapeutic drugs for DB-induced hepatotoxicity and the diverse biological activities of RRT, investigating whether RRTJ can ameliorate liver injury caused by DB and its potential mechanism is warranted. The results showed that 5 and 10 mL/kg RRTJ antagonized DB-induced liver injury, reduced serum ALT and AST levels, and improved pathological damage. Notably, 10 mL/kg RRTJ enhanced antioxidant enzyme activities (SOD, GSH-Px) and decreased lipid peroxidation product (MDA) levels. Further studies revealed that both doses of RRTJ can reduce the expression of autophagy-related proteins (Beclin1, LC3B, p62) caused by DB, suppress the ratio of LC3B II/I, and improve autophagic flux. Additionally, 5 and 10 mL/kg RRTJ can inhibit the activation of AMPK signaling induced by DB. This study is the first to investigate the role of autophagic flux impairment in DB-induced rat liver injury and the potential intervention of RRTJ at the animal level. (Figure 8) The effect of diosbulbin B on autophagic flux. The left panel shows normal autophagy, while the right panel indicates autophagic flux impairment induced by diosbulbin B.
Conclusion
The results demonstrated that treatment with DB led to oxidative damage and mitochondrial impairment in rats. Further studies revealed that DB administration resulted in autophagy dysfunction. Conversely, RRTJ intervention exhibited antioxidant properties, normalized autophagic activity, and mitigated DB-induced liver injury. Notably, this study has inherent limitations. Specifically, the limited sample size in this study may have affected the comprehensiveness and generalizability of the findings. Furthermore, this study did not utilize lysosomal markers (e.g., LAMP2, cathepsin) or autophagic flux assays (e.g., CQ/BAFA1 treatments combined with LC3 turnover analysis) to confirm autophagic flux obstruction. To address this limitation, future investigations could employ autophagic flux inhibitors (e.g., bafilomycin A1) or fluorescent lysosomal tracers (e.g., LysoTracker Red) to systematically characterize the interplay between autophagosome-lysosome fusion and substrate degradation. Additionally, it is strongly recommended to deploy siRNA-based gene silencing or knockout (KO) models to definitively establish the causal relationship among AMPK activation, autophagic regulation, and hepatoprotective outcomes. Such mechanistic validation would strengthen the demonstration of AMPK-mediated autophagy as a therapeutic target for liver protection
Supplemental Material
Supplemental Material - Rosa roxburghii tratt juice mitigates diosbulbin B-induced hepatotoxicity through autophagy regulation
Supplemental Material for Rosa roxburghii tratt juice mitigates diosbulbin B-induced hepatotoxicity through autophagy regulation by Yuhong Zhang, Xingcan Yang, Mao yuan Gong, Yang Yang, Ruobi Chen, Yuqi Li, Amin Wang, Yuyan Xu and Qibing Zeng in Human & Experimental Toxicology
Footnotes
Acknowledgments
The authors appreciate the help of Guizhou Medical University.
Ethical approval
The animal study was reviewed and approved by the Animal Ethics Committee of Guizhou Medical University, NO. 2303062. All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH) and adhered to the ARRIVE guidelines where applicable.
Informed consent
Consent for publication was obtained from each author.
Author contributions
All authors contributed to the study conception and design. Yuhong Zhang and Xingcan Yang: Conceptualization, methodology, data curation, visualization, writing—original draft preparation. Mao yuan Gong, Yang Yang, Ruobi Chen, Yuqi Li and Amin Wang: Methodology, data curation, visualization. Yuyan Xu: Conceptualization, resources, project administration, writing, reviewing, and editing. Qibing Zeng: Conceptualization, supervision, writing, reviewing, and editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by The Outstanding Young Talents Program of Guizhou Medical University (No. [2021]102), High–Level Talents Startup Fund Project of Guizhou Medical University (No. J [2020]075), and University-Level Key Laboratory Construction Project of Guizhou Medical University (No. the university-level key laboratory [2024]002).
Declaration of conflicting of interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets used and analyzed during this study are available from the corresponding author on reasonable request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.