
Abstract
Objective
To explore the differential expression of genes between wild-type chronic compressive injury (CCI) mice (WT-CCI) and interferon regulatory factors 4 (IRF4) knockout CCI mice (KO-CCI) by RNA-seq analysis of the mouse spinal cord.
Methods
RNA-seq analysis of the spinal cord tissue of the chronic sciatic nerve ligation mice and Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses were used.
Results
A total of 104 genes were up-regulated and 116 genes were down-regulated in spinal cord of the mice in IRF4 knockout (KO-CCI) group compared with that in the wild-type CCI (WT-CCI) group. There were 1472 differentially expressed genes in the biological process group, 62 differentially expressed genes in the cellular component group, and 163 differentially expressed genes in the molecular function group in KO-CCI mice. A total of 14 genes related to inflammatory reactions were differentially expressed. Real-time PCR results confirmed that Pparg and Grpr mRNA expression was up-regulated and Arg 1 and Ccl11 mRNA expression was down-regulated in the KO-CCI group.
Conclusion
IRF4 is involved in neuropathic pain in CCI mice, IRF4 may participate in neuropathic pain by regulating Grpr, Mas1, Galr3, Nos2, Arg1, Ccl11, Ptgs2, S100a8, Pparg, Cd40, Has2, Gpr151, Il123a, Capns2, Ankrd1, Ccnb1, and Nppb genes.
Introduction
Neuropathic pain is a type of chronic pain characterized by spontaneous pain, hyperalgesia, abnormal pain, and paresthesia.1,2 Its etiology includes physical mechanical injury, metabolic or nutritional neurological changes, viral infection, neurotoxicity of drugs or radiotherapy, ischemic neurological damage, neurotransmitter dysfunction, etc.3–8 Owing to the increased expression of Na+ channels and voltage-gated Ca2+ channels in the cell membrane of injured neurons, abnormal discharge of neurons causes spinal cord neurons to emit ectopic impulses, increases the sensitivity of spinal cord neurons and the transmission of neurotransmitters between synapses, changes the normal physiological activities of neurons, and intensifies the response to peripheral non-injurious or minor injury stimulation.9–11
Spinal microglia and inflammatory mediators are known to be closely associated with neuropathic pain.11–14 After nerve injury, the primary afferent end releases neurotransmitters that act on spinal glial cell receptors.
Interferon regulatory factors 4 (IRF4) was mainly expressed in immune cells and a key transcription factor for M2 macrophage polarization.15–24 IRF4 is involved in the regulation of microglial polarization and induces microglia to polarize toward M2. 25 As well as, IRF4 also mediates inflammation by up-regulating CCL17.26,27 Inflammatory reactions and mediators play an important role in neuropathic pain. IRF4 is a key factor in regulating the inflammatory response and mediates involvement in inflammatory pain.11–14 Therefore, we speculate that IRF4 may also participate in neuropathic pain by regulating inflammatory genes.
In this study, to investigate the role of IRF4 in neuropathic pain, transcriptomic analysis was performed to identify genes and pathways involved in IRF4 in neuropathic pain following chronic sciatic nerve ligation in mice.
Methods and materials
Animals and model preparation
This study obtained permission for the ethical review of animal welfare from the Experimental Animal Ethics Committee of Guangdong Medical Experimental Animal Center (No. C202108-9). Wild-type C57BL/6 mice (WT), weighing 20–22 g and 8 weeks old, were purchased from Zhuhai Baitantong Biotechnology Co., Ltd. (Zhuhai, China) and IRF4 knockout C57BL/6 mice (KO) were purchased from Shanghai Model Organisms Center, Inc. (Shanghai, China). In this study, Mice were anesthetized and chronic compressive injury (CCI) model were established. Due to the aim of this study to observe the expression of spinal cord-related genes in mice with chronic sciatic nerve ligation after IRF4 knockout, the analysis results of the differentially expressed genes were only compared between the mice in the WT-CCI group (native wild-type C57BL/6 CCI mice) and KO-CCI group (IRF4 knockout C57BL/6 CCI mice).
Mechanical withdrawal threshold (MWT)
There were eight mice in each group for the mechanical withdrawal threshold (MWT) detection. The MWT of mice was measured using a von Frey filament before surgery and 1, 3, 5, 10, 15, and 20 days after the operation, and the threshold value of 50% foot retraction was calculated by the up-down method with von Frey filament.
Total RNA extraction, transcriptome library construction and illumina sequencing
After the CCI model was established for 5 days, the spinal cord of the mice (three biological replicates in each group) was extracted. TRIzol reagent (Cat. No. 15596-018) was used to extract total RNA. A NanoDrop 2000 ultramicroanalyzer (Agilent Technologies, USA) was used to determine the RNA concentration, and an Agilent 2100 biological analyzer (Agilent Technologies, USA) was used to determine RNA integrity. The mRNA library was prepared according to the instructions of the VAHTS Universal V6 RNA-seq Library Prep Kit for Illumina (Cat. No. NR604). The mRNA was enriched with a poly A tail through VAHTS mRNA Capture Beads (Cat. No. N401), then randomly interrupt the mRNA obtained in the Frame/Prime Buffer. The double-stranded cDNA was synthesized and purified and then repaired. A tail was added, and a sequencing connector was connected. Approximately 370–420 bp of cDNA was screened using AMPure XP beans (Beckman Coulter, Cat. No. A63881), PCR amplification was performed, and the product purified using AMPure XP beans (Beckman Coulter, Cat. No. A63881) was used to obtain a library. After library construction, the Qubit 2.0 fluorescence quantizer (Thermo Fisher, USA) was used for preliminary quantification, which was diluted to 1.5 ng/µL, and the Agilent 2100 biological analyzer (Agilent Technologies, USA) was used to detect the insert size of the library.
Real-time PCR
The primer sequences of the detected genes.

Data analysis
The mechanical withdrawal threshold and mRNA expression were expressed as the Mean + SD and analyzed using SPSS 16.0, and the data were analyzed by paired t test, the test level was 0.05. Sequence data were removed from the connector sequence. We compared the filtered reads of each sample with the reference sequence (GRCm38 gencodevM25) using Star v2.7.1a software and quantified gene expression with FPKM as a unit using FeatureCounts v2.0.1 software.28,29 Differential expression analysis was performed using EdgeR v3.28.1 software. 30 In this analysis, the screening threshold for differential genes was set at p < .05 and | log2 (foldchange) |>1. Genes with | log2 (foldchange)>1 | were regarded as upregulated, and those with |log2 (foldchange) |<1 were regarded as downregulated. A heatmap cluster map of differentially expressed genes was generated using R software (R.3.6.0). The analysis software topGO v2.38.1 was used for GO enrichment analysis of differentially expressed genes, and Fisher's exact test was used for statistical analysis. The analysis software clusterProfiler v3.14.3 was used for KEGG pathway enrichment analysis of differentially expressed genes, and the statistical test method was the hypergeometric test. 31 The analysis software ClusterProfiler was used for GSEA of the GO and KEGG pathway datasets.
Results
Mechanical withdrawal threshold
After CCI surgery, the MWT of the mice in the two groups decreased significantly compared to that before surgery, reached the lowest value on the 5th day after surgery. Compared with the MWT of the mice in the WT-CCI group, it increased significantly in the KO-CCI group from the preoperative period to 20 days after the operation (Figure 1). The mechanical withdrawal threshold of the mice in the two groups (mean + SD, n = 8). *p < .05 versus that of the mice in the WT-CCI group.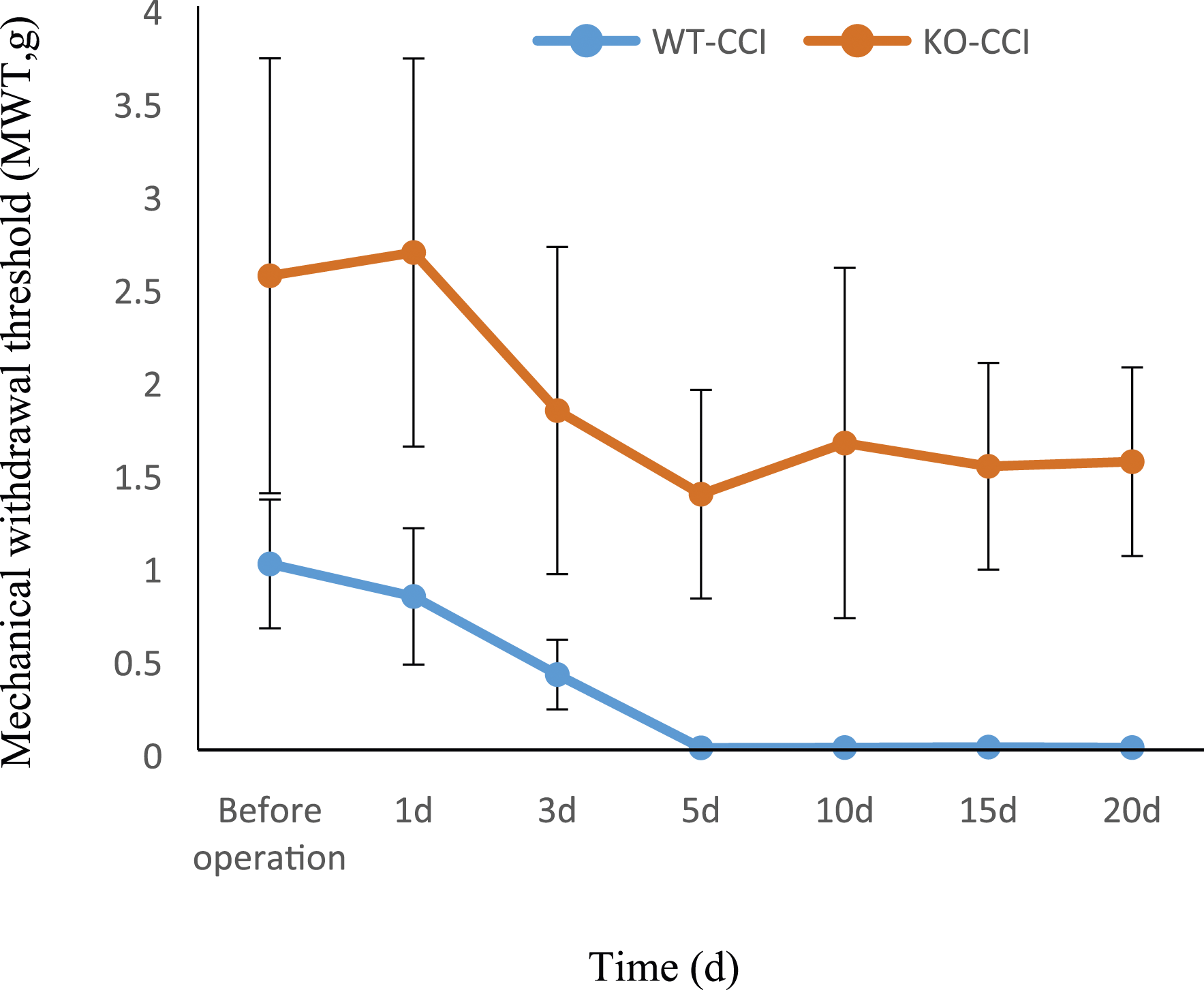
Sequencing and assembly
The reads mapping rate of each sample in those two groups.

Analysis of the differentially expressed genes (KO-CCI vs WT-CCI)
In this study, the screening criteria for differentially expressed genes were p value <.05 and | log2 (fold change) |>1. In total, 220 genes were differentially expressed. Compared to the WT-CCI group, 104 genes were significantly up-regulated and 116 genes were significantly down-regulated in the spinal cord of the KO-CCI group mice (Figure 2). Volcano plot of the differential gene expression of the spinal cord in mice in the KO-CCI group. There were 220 differentially expressed genes (p value <.05 and | log2 (fold change) |>1). The blue plot represents the down-regulated genes (116), the red plot represents the up-regulated genes (104), and the gray plot represents the genes with no differential expression.
Gene Ontology (GO) enrichment analysis (KO-CCI vs WT-CCI)
Gene Ontology (GO) is involved in Molecular Function (MF), Biological Process (BP), and Cellular Component (CC). The differentially expressed genes in the KO-CCI group were mainly enriched in the following GO pathways: regulation of cell promotion and response to external stimuli. Compared with those in the WT-CCI group, there were 1472 differentially expressed genes in the BP group, 62 differentially expressed genes in the CC group, and 163 differentially expressed genes in the MF group in the spinal cord of KO-CCI mice. We conducted GO enrichment analysis on the differentially expressed genes between the KO-CCI group and the WT-CCI group, with p < .01. GO functional enrichment analysis revealed that knocking out the IRF4 gene had an impact on the following CC: condensed chromosomes, central region, condensed chromosomes; MF: RAGE receiver binding, empirical extracellular ligand-gated, ion channel activity; BP: regulation of response to external stimuli, regulation of cell promotion, cell promotion, etc. (Figure 3). Gene enrichment analysis of the spinal cord in the KO-CCI group compared with the WT-CCI group. ((a): BP; (b) CC; (c) MF).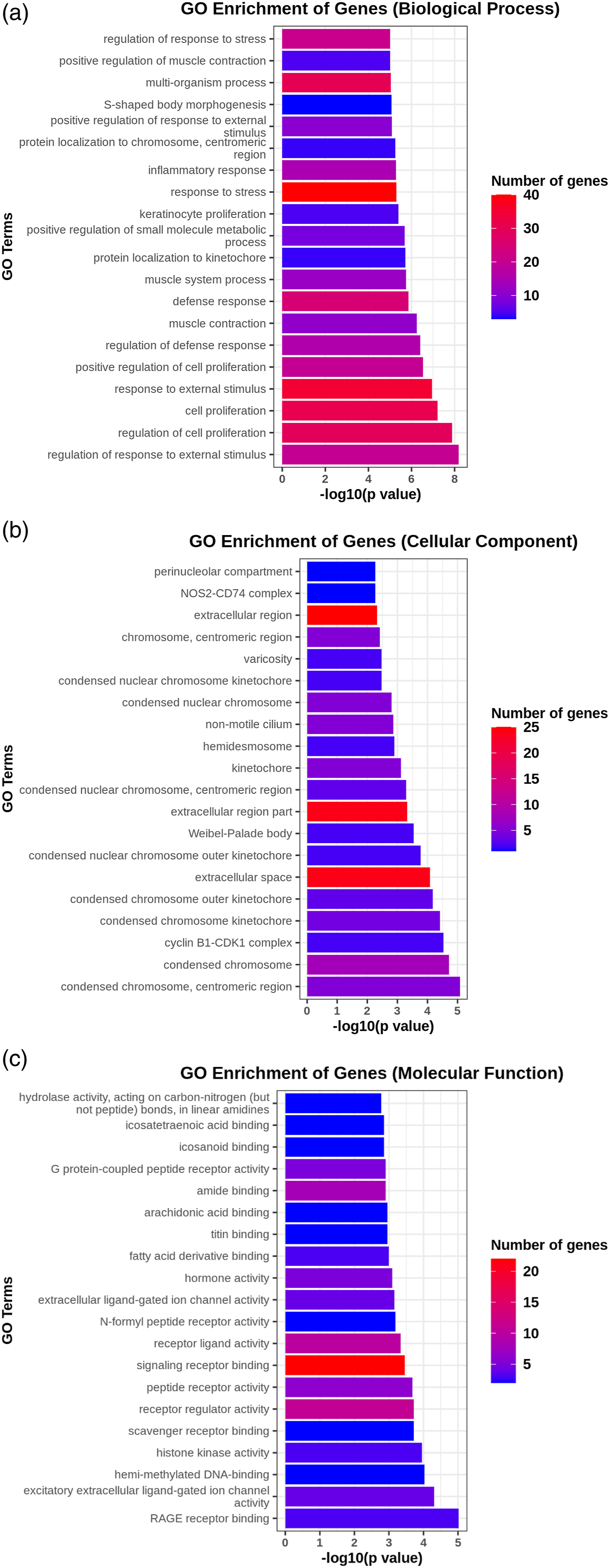
Kyoto encyclopedia of genes and genomes analysis of differential genes (KO-CCI vs. WT-CCI)
In this study, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG) database to perform KEGG pathway enrichment analysis of differentially expressed genes. Compared with those of the mice in the WT-CCI group, the differential genes of the mice in the KO-CCI group were enriched in neuroactive ligand‒receptor interaction (13 genes), arginine biosynthesis (2 genes), and the IL-17 signaling pathway (3 genes) (Figure 4). In Figure 4, the Rich Factor refers to the ratio of the number of differential genes enriched in the path to the number of annotated genes. Top 20 enriched pathways from KEGG analysis of differentially expressed genes (KO-CCI vs. WT-CCI).
Differential expression of inflammatory response-related genes (KO-CCI vs. WT-CCI)
Differential expression of inflammatory response-related genes in the spinal cord of mice in KO-CCI and WT-CCI groups.

Expression of some inflammatory response-related genes
There was no Irf4 mRNA expression in KO-CCI mice. Compared to the WT-CCI group, Pparg, Grpr, and Irf4 mRNA expression was up-regulated in the KO-CCI group. In addition, the Arg 1 and Ccl11 mRNA expression levels of the mice in the KO-CCI group were down-regulated compared with those in the WT-CCI group (Figure 5). Expression of some related genes (mean + SD, n = 3). *p < .01 versus that of the mice in the WT-CCI group. (a) Pparg mRNA expression, (b) Grpr mRNA expression, (c) Arg1 mRNA expression, (d) Irf4 mRNA expression, (e) Ccl11 mRNA expression.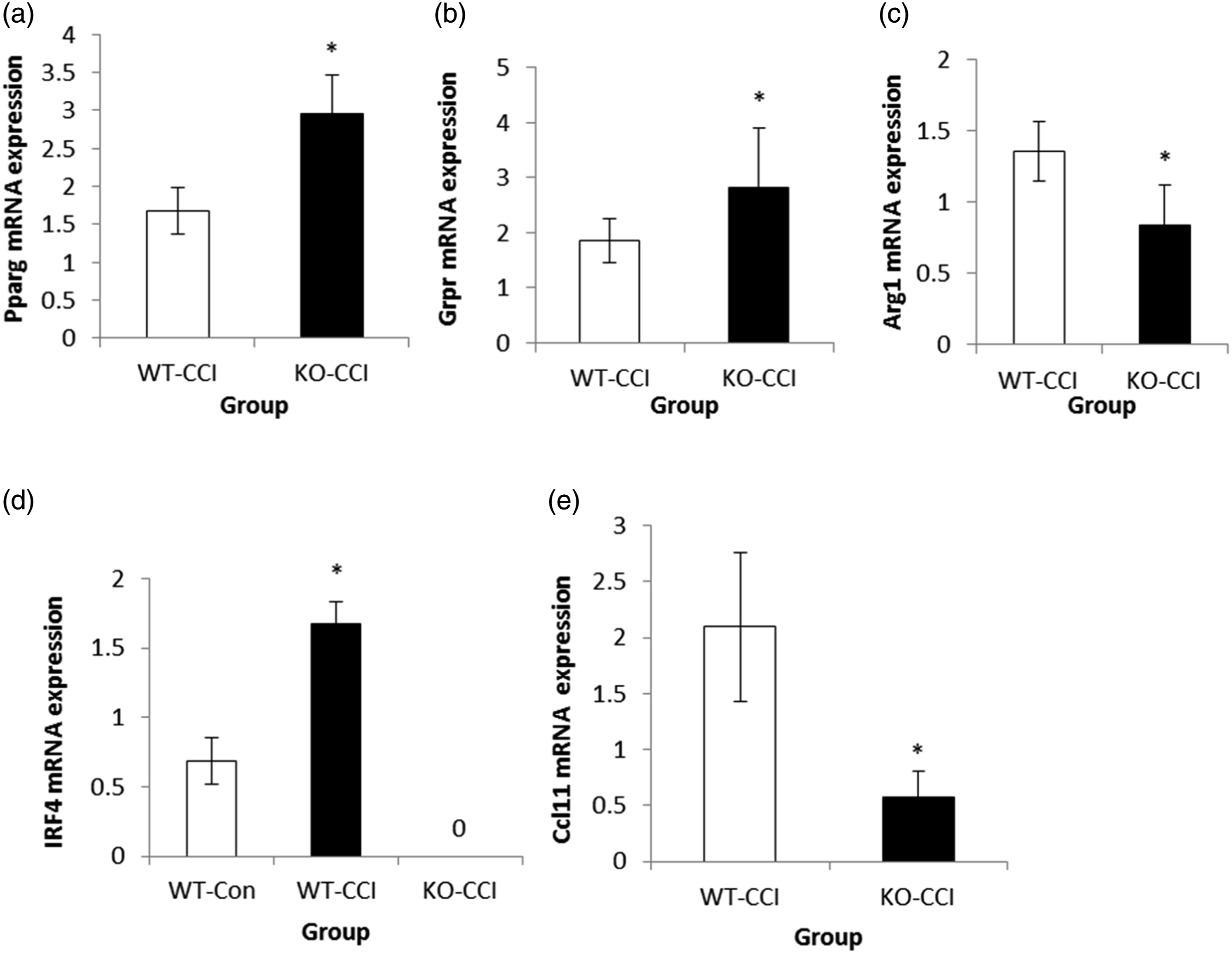
Discussion
Chronic neuropathic pain is a common clinical condition, and inflammation is an important cause of neuropathic pain.8,32,33 Anti-inflammatory cytokines have shown analgesic effects in various animal models of neuropathic pain. 32 The release of inflammatory mediators can lead to a wide range of immune responses, resulting in a neuroinflammatory environment, which causes the activation of glial cells, releases more media, and intensifies the inflammatory response in an uncontrolled manner. 34 IRF4 is an important regulator of the inflammatory response. It can up-regulate the inflammatory factor CCL17, produce proinflammatory effects, and mediates inflammation and inflammatory pain. 35
RNA sequencing has been widely used in the study of neuropathic pain.36,37 Some studies have confirmed that IRF4 was involved in neuropathic pain. Its downstream target, CCL17, has also been shown to be associated with pain.35,38 In this study, we used the spinal cord tissue of IRF4 knockout CCI (KO-CCI) mice and wild-type (WT-CCI) mice for RNA-Seq analysis to screen for differentially expressed genes. Compared to the WT-CCI group, 220 genes showed differential expression, 104 genes were significantly up-regulated, and 116 genes were significantly down-regulated in the spinal cord of the KO-CCI group.
Kyoto encyclopedia of genes and genomes enrichment pathway analysis revealed that the differentially expressed genes in the mice in the KO-CCI group. These genes are closely associated with the development of neuropathic pain. There were 13 genes involved in neuroactive ligand‒receptor interactions: Glra1,Grpr,Tbxa2r,Edn1,Lhb,P2rx1,Mas1,Galr3,Chrne,Ucn,Fpr1,Fpr2,Ghrl. Previous studies have shown that the gastrin-releasing peptide receptor (Grpr), 39 Spinal Mas1, 40 Galr3, 41 Nos2, 42 Arg1 43 ,Ccl11 and S100a8 44 were closely involved with neuropathic pain or inflammatory pain. KEGG enrichment pathway analysis and previous study results suggested that the mechanism of neuropathic pain in IRF4 knockout mice may be related to Grpr, Mas1, Galr3, Nos2, Arg1, Ccl11, Ptgs2, and S100a8. IRF4 may participate in neuropathic pain by regulating Grpr, Mas1, Galr3, Nos2, Arg1, Ccl11, Ptgs2, and S100a8.
We conducted differential analysis of the genes in the mice spinal cord in the KO-CCI group in the inflammatory response pathway. The results showed that the Pparg gene was up-regulated, and the Cd40, Has2, Gpr151, Il123a, Capns2, Ankrd1, Ccnb1, and Nppb genes were down-regulated. Based on the sequencing results of differentially expressed genes in the inflammatory response, we selected the Pparg, Grpr, Ccl11, and Arg1 genes for PCR detection to verify the transcriptome sequencing results. The PCR detection results were consistent with the transcriptome sequencing results.
Conclusion
Interferon regulatory factors 4 is involved in neuropathic pain in CCI mice, IRF4 may participate in neuropathic pain by regulating Grpr, Mas1, Galr3, Nos2, Arg1, Ccl11, Ptgs2, S100a8, Pparg, Cd40, Has2, Gpr151, Il123a, Capns2, Ankrd1, Ccnb1, and Nppb genes.
Footnotes
Author contributions
All the authors participated in the design and review of the manuscript. WX designed and implemented the experiments and wrote the paper. PJ, EQ, WY, and ZZ implemented the experiments, collected the data, and performed the statistical analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Scientific Research Project of Guangdong Administration of Traditional Chinese Medicine (No. 20231323), the Guangdong Medical Science and Technology Research Foundation (No. A2022341), and the Hospital Level Project of Foshan Second People's Hospital (No. A2022A01).
Data Availability Statement
If needed, data can be provided by WX.