
Abstract
Objective
The ability of glutathione S-transferase zeta 1 (GSTZ1) to modulate homeostasis of cellular redox and induce ferroptosis was explored in bladder cancer cells, and the involvement of the high mobility group protein 1/glutathione peroxidase 4 (HMGB1/GPX4) in these effects was studied.
Methods
BIU-87 cells stably overexpressing GSTZ1 were transfected with appropriate plasmids to deplete HMGB1 or overexpress GPX4, then treated with deferoxamine and ferrostatin-1. Antiproliferative effects were assessed by quantifying levels of ferroptosis markers, such as iron, glutathione (GSH), malondialdehyde (MDA), reactive oxygen species (ROS), GPX4, transferrin, and ferritin.
Results
GSTZ1 was significantly downregulated in bladder cancer cells. GSTZ1 overexpression downregulated GPX4 and GSH, while greatly increasing levels of iron, MDA, ROS, and transferrin. GSTZ1 overexpression also decreased proliferation of BIU-87 cells and activated HMGB1/GPX4 signaling. The effects of GSTZ1 on ferroptosis and proliferation were antagonized by HMGB1 knockdown or GPX4 overexpression.
Conclusion
GSTZ1 induces ferroptotic cell death and alters cellular redox homeostasis in bladder cancer cells, and these effects involve activation of the HMGB1/GPX4 axis.
Introduction
Glutathione S-transferase zeta 1 (GSTZ1) polymorphisms are associated with increased risk of bladder cancer. 1 GSTZ1 is a member of the glutathione S-transferase (GST) family, which catabolizes phenylalanine and modulates cellular redox homeostasis by repressing nuclear factor erythroid 2-related factor 2 (NRF2). 2 In addition, ferroptosis is characterized by build-up of reactive oxygen species (ROS) and iron-dependent accumulation of lipid peroxide intracellularly.3,4 Ferroptosis dowenregulation is closely associated with the occurrence and development of various tumors, including bladder cancer. 5
Moreover, GSTZ1 can trigger ferroptosis by directly repressing the cell surface cysteine-glutamate antiporter (system Xc-), decreasing the cellular antioxidant glutathione (GSH). 6 Phospholipid hydroperoxide glutathione peroxidase 4 (GPX4) modulates ferroptotic cancer cell death: it is inhibited during ferroptosis, and overexpressing it inhibits ferroptosis.7,8 Suppressing ferroptosis may be a therapeutic strategy against bladder cancer. 1
GST levels are positively associated with expression of high-mobility group box-1 (HMGB1) in some diseases.9,10 HMGB1 acts via the NRF2/GPX4 axis to repress ferroptosis in mesangial cells in response to high glucose. 11 HMGB1 has also been shown to induce ferroptosis via the RAS-JNK/p38 signaling pathway in leukemia. 12 Whether GSTZ1 activates ferroptosis by altering HMGB1 expression is unclear.
In the present study, we began to explore how GSTZ1 drives ferroptosis in bladder cancer cells. Our assays suggest that GSTZ1 acts via the HMGB1/GPX4 axis to increase ROS levels and intracellular iron pools, thereby triggering ferroptosis of cancer cells.
Material and methods
Cell culture and treatment
Human bladder cancer cell lines RT4 and BIU-87 as well as the human uroepithelial cell line SV-HUC-1 were purchased from the American Type Culture Collection (Manassas, VA, USA). These cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco BRL, Rockville, MD, USA) supplemented with 10% fetal bovine serum at 37°C in an atmosphere containing 5% CO2 at 95% relative humidity. BIU-87 cells were treated with the indicated concentrations of deferoxamine, ferrostatin-1, necrostatin-1, chloroquine, or Z-VAD-FMK for 24 h. All drugs were purchased from Selleck Chemical (Houston, TX, USA). 5
Cell transfection
The following six pcDNA3.1 plasmids were synthesized by Biofavor (Wuhan, China): empty pcDNA3.1 vector (vector), plasmid overexpressing human GSTZ1 (OE-GSTZ1), plasmid overexpressing human HMGB1 (HMGB1), plasmid encoding human GPX4 (GPX4), plasmid encoding a short interfering RNA against HMGB1 (RNAi-MGB1), and plasmid encoding short interfering RNA as a negative control shRNA (RNAi-sh). BIU-87 cells were transfected for 24 h using Lipofectamine 2000 (Nikon, Tokyo, Japan) according to the manufacturer’s instructions.
Cell viability
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8, MedChemExpress, Monmouth Junction, NJ, USA) based on the manufacturer’s instructions. Briefly, BIU-87 cells were grown overnight in 96-well, flat-bottom microtiter plates (5 ×103 cells/well). After transfection or treatment for 24 h, 10 μL of CCK-8 solution was added to each well and incubated for 2 h. Absorbance at 450 nm was measured on a microplate reader (Synergy HT, Bio-Tek, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
RT4, BIU-87, and SV-HUC-1 cell lines were cultured in 6-well plates (1 ×105 cells/well) for 48 h. Total RNA was isolated from cells using TRIzol reagent (BD Pharmingen, Shanghai, China) according to the manufacturer’s instructions. The purified RNA was reverse-transcribed into cDNA using the PrimeScript™ RT Reagent Kit (Santa Cruz Biotechnology, USA). Primers were as follows: GSTZ1 forward, 5′-CTACTTCTGCATATGGCGAATTCCGGC-3'; GSTZ1 reverse, 5′-GATGATGATGGGCGGCCGCGATGGTGGA-3'; β-actin forward, 5′-CATGTACGTTGCTATCCAGGC-3'; and β-actin reverse, 5′-CATGTACGTTGCTATCCAGGC-3'. Levels of mRNA were quantified using the SYBR Green qPCR Master Mix (Invitrogen, Thermo Fisher Scientific, USA). Levels of target mRNAs were calculated using the 2–ΔΔCt method and normalized to the level of β-actin. Each sample was analyzed in triplicate. 11
Western blotting
Cells were collected and lysed with RIPA buffer. The concentration of total soluble protein in each sample was measured using a BCA Protein Assay Kit (Sungene Biotech, Tianjin, China). Aliquots of soluble protein were mixed with denaturing loading buffer and boiled for 5 min at 100°C. Then 50 μg of protein from each sample was fractionated on a 12% SDS-PAGE gel, and transferred through the wet-transfer system onto polyethylene membranes. Non-specific binding sites were blocked with 5% skim milk at room temperature for 1 h. Membranes were washed three times with Tris-buffered saline containing 0.2% Tween-20 (TBST), then incubated at 4°C overnight with primary antibodies against GSTZ1 (catalog no. ab153995, Abcam, UK), HMGB1 (ab18256), GPX4 (ab125066), transferrin (ab214039), ferritin (ab75973), or β-actin (ab7817). All primary antibodies were diluted 1:1000. On the following day, membranes were washed with TBST, then incubated with horseradish peroxidase-conjugated IgG secondary antibody at room temperature. Protein bands were visualized using enhanced chemiluminescence (Bio-Tek) and quantified using Image J 4.0 (US National Institutes of Health, Bethesda, MD, USA). 12
Iron, glutathione (GSH), and malondialdehyde (MDA) assays
Treated BIU-87 cells were harvested into phosphate-buffered saline using a cell spatula and lysed in 1% Triton (Bio-Tek) on ice for 30 min. Lysates were centrifuged at 12,000 g and 4°C for 12 min. The supernatant was assayed for iron, GSH, and MDA using commercial kits (Guangzhou RiboBio, Guangzhou, China) following the manufacturer’s instructions. 12
ROS assay
Following transfection or treatment, BIU-87 cells (1 ×105 cells/well) were cultured in serum-free DMEM in 6-well plates. Cells were treated in the dark for 30 min at 37°C with 10 μM of DCFH-DA (Beyotime, Shanghai, China), which was added directly to the culture medium. Cells were rinsed three times with serum-free medium, then analyzed by flow cytometry (BD, Franklin Lakes, NJ, USA).
Statistical analysis
Results were shown as mean ± standard deviation (SD) from three independent experiments. Statistical analysis was performed using Prim 7.0c GraphPad Software (GraphPad Software, Inc., USA). Differences between groups were assessed using t-test (two groups) or one-way ANOVA (multiple groups). p < 0.05 was regarded as statistically significant.
Results
GSTZ1 upregulates ferroptotic mediators in bladder cancer cells
Levels of GSTZ1 mRNA and protein were significantly lower in both bladder cancer cell lines than in healthy uroepithelial cells (Figures 1(a) and (b)). Degradation of the iron storage protein ferritin and accumulation of the iron uptake protein transferrin contribute to the production of ROS, which then triggers ferroptosis.13,14 Therefore we examined whether GSTZ1 modulates the expression of proteins that regulate iron accumulation and redox processes. Overexpressing GSTZ1 in BIU-87 cancer cells downregulated GPX4 while upregulating transferrin, without affecting ferritin levels (Figure 1(c)). High expression of GSTZ1 also upregulated iron, MDA, and ROS while downregulating GSH and reducing cell viability (Figures 1(d)–(h)). These results suggest that GSTZ1 inhibits the proliferation of bladder cancer cells at least in part by stimulating ferroptosis. GSTZ1 overexpression increases reactive oxygen species and iron accumulation in bladder cancer cells. (a) Levels of GSTZ1 mRNA in healthy control (SV-HUC-1) or bladder cancer cell lines (RT4 and BIU-87), based on qRT-PCR. (b) Levels of GSTZ1 protein, based on western blotting and densitometry. (c) Western blot and densitometry of GSTZ1, GPX4, transferrin, and ferritin in BIU-87 cells overexpressing GSTZ1. (d–g) Levels of (d) iron, (e) glutathione (GSH), (f) malondialdehyde (MDA), and (g) reactive oxygen species in BIU-87 cells overexpressing GSTZ1. (h) Cell viability was determined using the CCK-8 assay. Data are mean ± SD (n = 3). *p < 0.05 vs control.
Overexpression of GSTZ1 triggers ferroptosis in bladder cancer cells
In order to assess the role of GSTZ1 in triggering cell death, BIU-87 cells overexpressing GSTZ1 were treated with inhibitors of cell death (Figure 2). Only treatment with the iron chelator deferoxamine or ferrostatin-1, a potent inhibitor of ferroptosis, reversed the GSTZ1-mediated decrease in cell viability and increases in iron, GSH, MDA, and ROS. These results confirm that GSTZ1 inhibits the proliferation of bladder cancer cells at least in part by stimulating ferroptosis. Deferoxamine and ferrostatin-1 antagonize the effects of GSTZ1 overexpression in bladder cancer cells. BIU-87 cells overexpressing GSTZ1 (OE-GSTZ1) were treated with 100 μM deferoxamine (DOM), 1 μM ferrostatin-1 (Fer-1), 10 μM necrostatin-1 (Nec-1), 25 μM chloroquine (CHQ) or 10 μM Z-VAD-FMK for 24 h. (A–D) Levels of (a) iron, (b) glutathione (GSH), (c) malondialdehyde (MDA), and (d) reactive oxygen species (ROS) were assayed. (e) Cell viability was determined using the CCK-8 assay. Data are mean ± SD (n = 3). *p < 0.05 vs control; #p < 0.05 vs OE-GSTZ1.
GSTZ1 triggers ferroptosis in bladder cancer cells by activating the HMGB1/GPX4 axis
HMGB1 downregulates GPX4 to induce ferroptosis in damaged neuronal cells.
15
Thus, we examined whether GSTZ1 acts via the HMGB1/GPX4 signaling axis to stimulate ferroptosis in BIU-87 cells. GSTZ1 overexpression upregulated HMGB1 and downregulated GPX4, whereas GSTZ1 knockdown had the opposite effects (Figure 3(a) and (b)). GSTZ1 overexpression was associated with higher levels of iron, MDA, and ROS, as well as lower GSH levels and cell viability (Figure 3(c)–(g)). These results suggest that GSTZ1 induces ferroptosis in BIU-87 cells through the HMGB1/GPX4 axis. GSTZ1 modulates HMGB1/GPX4 signaling in BIU-87 cells. (a) Levels of GSTZ1, HMGB1, and GPX4 in BIU-87 cells overexpressing GSTZ1 (OE-GSTZ1), as detected by western blot. (b) Levels of GSTZ1, HMGB1, and GPX4 in BIU-87 cells in which endogenous GSTZ1 was knocked down using RNA interference (RNAi-GSTZ1). Control cells were transfected with negative control RNA (RNAi-sh). (c–f) Levels of (c) iron, (d) glutathione (GSH), (e) malondialdehyde (MDA), and (f) reactive oxygen species (ROS). (e) Cell viability was determined using the CCK-8 assay. Data are mean ± SD (n = 3). *p < 0.05 vs control.
GSTZ1 induces ferroptosis in bladder cancer cells by upregulating HMGB1
To explore the role of HMGB1 in GSTZ1-induced ferroptosis, HMGB1 expression was knocked down in bladder cancer cells overexpressing GSTZ1. Overexpression of GSTZ1 upregulated HMGB1 and transferrin, while downregulating GPX4; HMGB1 depletion antagonized these effects (Figure 4(a)). Ferritin levels did not depend on levels of GSTZ1 or HMGB1. These results suggest that GSTZ1 acts upstream of the HMGB1/GPX4 axis. HMGB1 downregulation antagonizes GSTZ1-induced ferroptosis in bladder cancer cells. BIU-87 cells stably overexpressing GSTZ1 (OE-GSTZ1) were transfected for 24 h with negative-control short interfering RNA (RNA-sh) or short interfering RNA targeting HMGB1 (RNAi-HMGB1). (a) Western blot of GSTZ1, HMGB1, GPX4, transferrin, and ferritin. (b–e) Levels of (b) iron, (c) glutathione (GSH), (d) malondialdehyde (MDA), and (e) reactive oxygen species (ROS) were assayed. (f) Cell viability was determined using the CCK-8 assay. Data are mean ± SD (n = 3). *p < 0.05 vs control; #p < 0.05 vs OE-GSTZ1.
Consistently, GSTZ1 overexpression significantly upregulated iron, MDA, and ROS levels, while reducing GSH levels and cell viability; knocking down HMGB1 in these cells partially reversed these effects (Figure 4(b)–(f)). Conversely, overexpressing both GSTZ1 and HMGB1 led to greater ferroptosis and lower viability than with GSTZ1 overexpression alone (Figure 5(a)–(e)). HMGB1 up-regulation promoted GSTZ1-induced ferroptosis in BIU-87 cells. BIU-87cells stably over-expressing GSTZ1 were transfected with empty vector (Vector) or a plasmid over-expressing HMGB1 (HMGB1) for 24 h. (a–d) Levels of (a) iron, (b) glutathione (GSH), and (c) malondialdehyde (MDA), and (d) relative intracellular levels of reactive oxygen species (ROS) in BIU-87 cells were measured. (e) Cell viability was determined by CCK-8 assay. Data are mean ± SD (n = 3). *p < 0.05 vs control; #p < 0.05 vs OE-GSTZ1.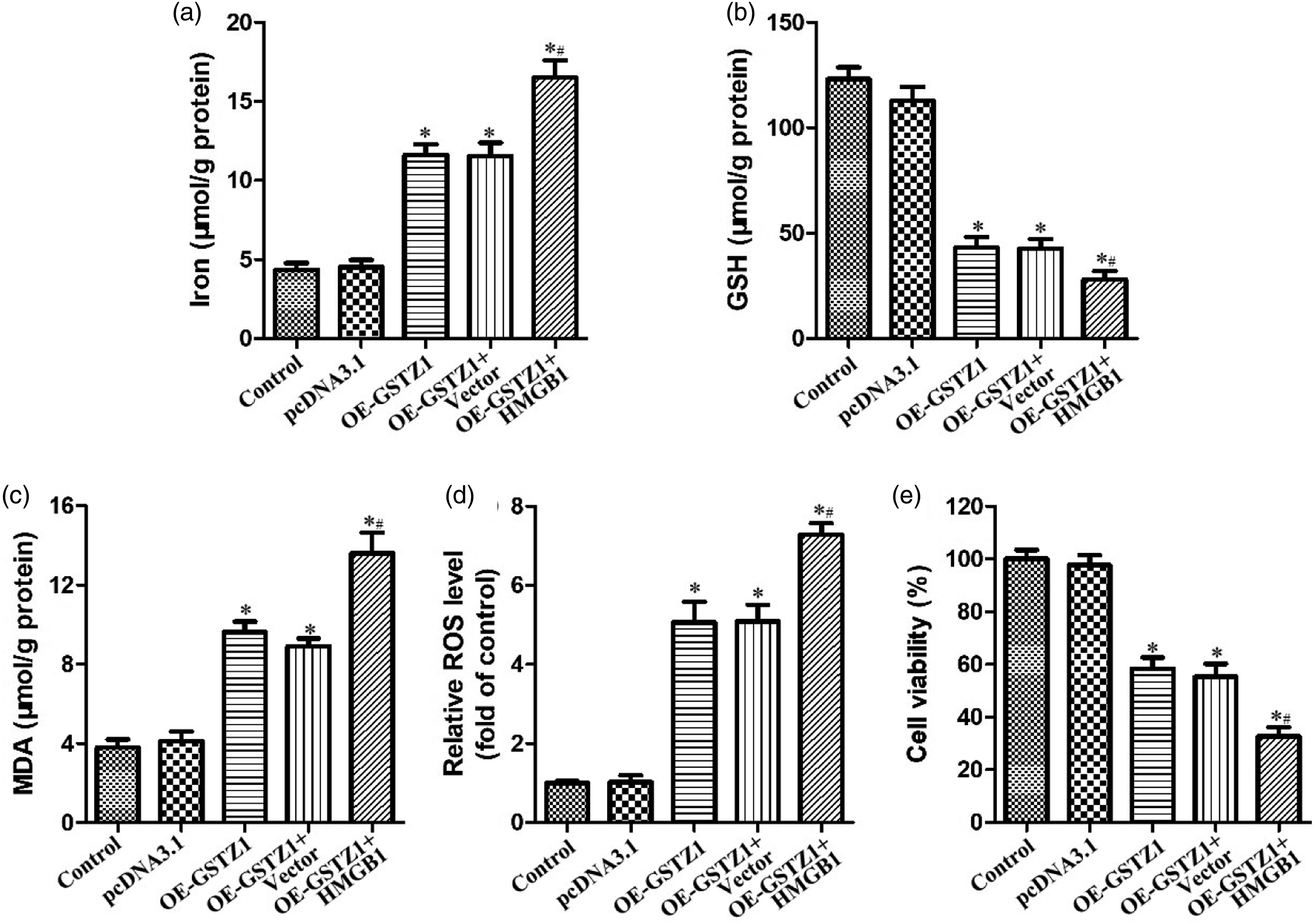
Taken together, our findings suggest that GSTZ1 induces ferroptosis by activating the HMGB1/GPX4 axis in bladder cancer cells, and that inhibition of this axis can antagonize the antitumor activity of GSTZ1.
GPX4 overexpression antagonizes GSTZ1-induced ferroptosis in bladder cancer cells
GPX4, a key regulator of ferroptosis, is inhibited by GSTZ1 in hepatocellular carcinoma cells,
7
and here we showed that GPX4 is also inhibited in bladder cancer cells overexpressing GSTZ1. Overexpressing GPX4 in these cells reversed the effects of GSTZ1, including the effects on levels of iron, MDA, and ROS (Figure 6). These results suggest that the ability of GSTZ1 to induce ferroptosis in bladder cancer cells requires GPX4 downregulation. GPX4 overexpression antagonizes GSTZ1-induced ferroptosis in bladder cancer cells. BIU-87 cells overexpressing GSTZ1 (OE-GSTZ1) were transfected with empty vector (Vector) or pcDNA3.1-GPX4 (GPX4) for 24 h. (a) Levels of GSTZ1, HMGB1, GPX4, transferrin, and ferritin were measured using western blotting. (b–e) Levels of (b) iron, (c) glutathione (GSH), (d) malondialdehyde (MDA), and (e) reactive oxygen species (ROS) were assayed. (f) Cell viability was determined using the CCK-8 assay. Data are mean ± SD (n = 3). *p < 0.05 vs control; #p < 0.05 vs OE-GSTZ1.
Discussion
Bladder cancer is the ninth most common cancer in the world and the most common malignant tumor in the urinary system.6,16 Thus, comprehensive understanding of bladder cancer onset may lead to more effective treatments. We found that bladder cancer cells downregulate GSTZ1 and thereby reduce ferroptosis, implying that upregulating GSTZ1 may be a therapeutic strategy, which we confirmed here in vitro. In addition, we identified that the HMGB1/GPX4 axis was modulated by GSTZ1 to promote ferroptosis. Our insights may help guide the development of more effective treatments against bladder cancer.
We explored ferroptosis in bladder cancer because this type of cell death has been closely associated with the occurrence and development of tumors, and induction of ferroptosis has emerged as a new type of cancer treatment.17,18 Numerous studies have explored how to stimulate ferroptosis by altering GPX4 expression or intracellular iron homeostasis in cancers affecting lung, clear cell renal cells, endometrium, colorectum, and bladder. For example, GPX4-mediated ferroptosis was induced by miR-324-3p to reduce cisplatin resistance in lung adenocarcinoma cells; 19 suppressor of variegation 3-9 homolog 1 (SUV39H1) depletion represses the proliferation of clear cell renal cell carcinoma though the ferroptosis induction by reducing GPX4; 20 PTPN18 downregulation triggered ferroptosis in endometrial cancer cells by modulating p-P38-mediated GPX4/xCT inactivation. 21 In addition, some drugs, including apatinib and bupivacaine have also been reported to modulate ferroptosis to induce the death of cancer cells.22,23 Moreover, lncRNA RP11-89 could facilitate tumorigenesis and ferroptosis resistance through PROM2-activated iron export by sponging miR-129-5p in bladder cancer. 24 Our study also suggests that upregulating ferroptosis may also be effective against bladder cancer, and we identified several molecules through which ferroptosis can be influenced, including GSTZ1, HMGB1 and GPX4.
Anticancer drugs can induce ferroptosis by upregulating GSTs.25,26 The GST GSTZ1 has been shown to repress NRF2/GPX4 signaling in hepatocellular carcinoma cells, potentiating the ability of sorafenib to trigger ferroptosis. 7 GPX4-dependent glutathione high-consumption drives acquired platinum chemoresistance in lung cancer-derived brain metastasis and liver precancerous lesion.27,28 GSTZ1 variants of lower activity may increase risk of bladder cancer by failing to metabolize haloacetic acids.29,30 We found here that bladder cancer cells downregulate GSTZ1, presumably to prevent ferroptosis from slowing tumor growth. We also found that upregulating GSTZ1 can induce ferroptosis and tumor cell death.
HMGB1 has been shown to slow progression of bladder cancer and to increase ROS accumulation in cancer cells, triggering ferroptotic death.7,31 We found that GSTZ1 induces ferroptosis in bladder cancer cells by upregulating HMGB1 and downregulating GPX4. HMGB1 depletion or GPX4 overexpression negated the effects of GSTZ1, and HMGB1 depletion also reversed GSTZ1-induced downregulation of GPX4, as shown in that GSTZ1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma cells by repressing NRF2/GPX4 pathway. 7 Furthermore, previous reports have revealed the potential applications of ferroptosis in the context of systemic therapy, radiotherapy and immunotherapy.23,32 Our study highlights that GSTZ1 induces ferroptotic cell death and alters cellular redox homeostasis in bladder cancer cells, and these effects may provide a therapeutic strategy for bladder cancer.
Conclusions
In summary, GSTZ1 upregulation contributes to ferroptosis in bladder cancer via repressing HMGB1/GPX4 axis; thereby inhibiting this signaling pathway may exert a therapeutic benefit in bladder cancer cases with GSTZ1 low-expression. The present observation also provide new insights into the molecular basis of the role of GSTZ1 in ferroptosis and proliferation of cancer cells. Therefore, our study identifies ferroptosis generally and the GSTZ1/HMGB1/GPX4 axis specifically as therapeutic targets in bladder cancer, and it provides new insights into the role of GSTZ1 in ferroptosis-induced cell death more broadly.
Footnotes
Acknowledgements
The authors thank the Xiangyang NO.1 People’s Hospital.
Author contributions
HYZ and QTC conceived and designed the research. LLZ conducted the experiments. YZ analyzed and interpreted the data. HYZ drafted the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.