
Abstract
Sepsis-associated encephalopathy (SAE) is a cognitive impairment caused by sepsis, associated with increased morbidity and death. And acetaminophen (APAP) is a promising therapeutic medicine for SAE treatment. This research was designed to determine whether APAP alleviates SAE by attenuating ferroptosis and mediating the glutathione peroxidase (GPX4) pathway. The cecal ligation and puncture (CLP) approach was used to establish septic mouse models. The survival rates for 7 days were determined. The Morris water maze (MWM) was utilized to assess cognitive function. Hematoxylin and eosin (HE) staining identified histopathologic alterations in hippocampal tissue. Mitochondrial damage was discovered in hippocampal tissue using transmission electron microscopy (TEM). The reactive oxygen (ROS) levels in hippocampal tissue were measured using commercial assays. Septic cell models were produced using HT22 cells grown with 1 μg/ml lipopolysaccharide (LPS). ROS were quantified using immunofluorescence. Ferroptosis-related protein expression levels in hippocampal tissue and HT22 cells were measured using western blotting. To evaluate the iron content of hippocampal tissue and HT22 cells, commercial kits were employed. According to the findings, APAP improved survival rates, lowered hippocampal and mitochondrial damage, and improve cognitive impairment. In both animal and cell studies, APAP reduced iron content, ROS, glutamate antiporter (xCT), 4-hydroxy-2-nonenal (4-HNE) levels but increased GPX4 expression. However, RSL3, a GPX4 inhibitor that acts as a ferroptosis activator, decreased the protective properties of APAP in vitro. Our findings suggest that APAP reduces sepsis-induced cognitive impairment by reducing ferroptosis, which is mediated by the GPX4 signaling pathway.
Background
Sepsis-associated encephalopathy (SAE) is a kind of cognitive impairment and acute central nervous system dysfunction induced by a systemic infection that is associated with higher morbidity and mortality in the intensive care unit (ICU).1,2 However, the pathophysiology of SAE is complicated and not completely understood. Medical research has focused on finding the likely pathogenesis of SAE and the feasible therapeutic possibilities.
Previous research has focused on the role of inflammatory and oxidative stress in SAE; however, therapeutic techniques targeting inflammatory and oxidative stress have not significantly reduced SAE. 3 In recent years, there has been growing interest in the function of trace elements in the course of sepsis. 4 Iron, an essential component for life, is required by the host and microbes to keep life functioning. 5 Clinical and scientific studies have demonstrated that in septic patients, lower blood iron levels, greater lipid peroxide levels, and iron metabolic anomalies are closely associated with an elevated risk of distant morbidity and death, as well as of cognitive impairment. 6 To isolate iron from microbes to limit their survival, the organism transfers significant amounts of iron into the cells, resulting in intracellular iron overload and the Fenton reaction, which results in cellular ferroptosis—a recently discovered mode of cell death caused by iron-dependent lipid oxidative damage. 7 Unlike apoptosis, necrosis, and autophagy, ferroptosis is characterized by decreased cell volume and increased mitochondrial membrane density without the characteristic apoptosis and necrotic manifestations.8,9 Furthermore, the brain, which has a quick energy metabolism and is high in polyunsaturated fatty acids, is prone to ferroptosis because it has a limited ability to scavenge ROS and antioxidants on its own.10,11 Some studies have recently revealed that hippocampal neuronal ferroptosis may be associated with the occurrence and development of brain damage and long-term memory impairment induced by ischemia or infection.12,13
Acetaminophen (APAP) is a commonly used antipyretic and analgesic drug. Low-dose APAP protects against mitochondrial toxicity by scavenging brain ROS, decreasing brain lipid peroxidation, boosting brain microsomal GSH-Px activity, and decreasing the inflammatory response.14–16 These APAP actions are consistent with anti-brain ferroptosis. According to recent research, low-dose APAP, which reduces LPS-induced neuroinflammation and improves cognitive impairment in mice by lowering hippocampal neuronal death, might be a potential therapeutic option for SAE. 17 The underlying process, however, remains unknown.
This study aimed to determine how APAP affects hippocampal neuronal ferroptosis in septic brains and to examine other ways in which APAP reduces SAE.
Materials and methods
Animals
Healthy male C57BL/6J mice, weighing 22–24 g, 6 weeks old, were purchased from Tianyao Biotechnology Company (Tianjin, China) and housed in an environment free of specific pathogenic bacteria: temperature 22–24°C, relative humidity 50%–70%, alternating day and night every 12 h, and free access to water.
The Laboratory Animal Management Committee approved this study of Tianjin Medical University (approval no. IRB2020-DW-07), and the animal disposal process was conducted according to ethical requirements.
Cecal ligation and puncture model
The SAE model was created utilizing a modified cecal ligation and perforation method based on Jelinek et al.’s study. 18 Under 2% pentobarbital sodium solution (50 mg/kg) deep anesthesia, the skin and abdominal wall were incised along the midline of the abdomen for approximately 1 cm. The cecum was ligated below the ileocaecal flap near the 1/4th of the cecum end using a sterile 20 G needle at the distal end of the cecum near the ligature. The intestinal contents were extruded for approximately 0.2 mL. The ligated cecum was returned to the abdominal cavity and the intestinal contents, and the incision was sutured layer by layer. Postoperative fluid resuscitation was performed with subcutaneous saline injection (5 mL/100 g) into the neck. Subcutaneous antibiotics (meropenem, 25 mg/kg, once per day) were started 6 h after intraperitoneal injection for 3 days.
Hippocampal neuronal cell line (HT22) culture
HT22 cells (Cat# CL-0595, Punuosai, Wuhan, China) were cultured as apposed cells in high glucose-modified Dulbecco’s Eagle’s medium (DMEM, Cat# D6429, Sigma, USA) containing 10% fetal bovine serum (FBS, Cat# 12,103C, Sigma, USA) and 1% penicillin/streptomycin solution (Cat# TMS-AB2, Sigma, USA). Incubate at 37°C with 5% CO2. Cells with more than 80% growth density were digested with trypsin, and the second and third generations were used to be tested.
Experimental design
For animal research, animals were randomly assigned to 4 groups: sham group, sham + acetaminophen (APAP) group, CLP group, and CLP + APAP group. The septic mice were reproduced by cecal ligation and puncture (CLP) surgery in the CLP and CLP + APAP groups. Mice in the sham or sham + APAP groups only accepted laparotomies without ligation and puncture. At 1 h before the sham or CLP procedure, mice in the sham + APAP and CLP + APAP groups received 100 mg/kg/day APAP (in 200 μl sterile saline) through intraperitoneal injection until the conclusion of the trial, whereas the sham and CLP groups received an identical amount (200 μl) of sterile saline only. At 24 h following the sham or CLP procedure, some mice in each group were sacrificed and perfused for brain tissue collection, while the rest (n = 20 per group) were randomly selected to observe survival rates for a total of 7 days. At 4–10 days postoperatively, mice in each group (n = 10 per group) were subjected to the Morris water maze (MWM) test. The hippocampal tissue (n = 6 per group) in each group was selected for GPX4, xCT, and 4-HNE protein determination by western blot, for pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6) and HMGB1 measurement by enzyme-linked immunosorbent assay (ELISA), and for ROS and iron content detection by commercial kits. Brain slices (n = 3 per group) and ultrathin hippocampal sections (n = 3 per group) were used for morphological change observation and ultrastructural investigation, respectively (Figure 1(a)). Experimental design. (a) Male C57/6J mice (age 6–8 weeks; weight 20–25 g) received an intraperitoneal injection of 100 mg/kg APAP or an equivalent amount of normal saline 1 h before the sham or CLP procedure. Brain tissue was taken from different groups for testing 24 h following the sham or CLP procedure. Until the completion of the test, the surviving mice were given an intraperitoneal injection of 100 mg/kg/day APAP or an equivalent amount of normal saline. The Morris water assignment was completed from the 4th to the 10th days following the sham or CLP procedure. (b) The mouse HT22 cell lines were cultured in control media, APAP medium, LPS medium, RSL3 medium, LPS+APAP medium, APAP + RSL3 medium, LPS+RSL3 medium, and LPS+APAP+RSL3 medium. The cells and culture medium supernatant were collected for the test at 24 h after incubation. CLP, cecal ligation and puncture; LPS, lipopolysaccharide; APAP, acetaminophen; RSL3, a GPX4 inhibitor.
In the cell experiment, RSL3, a GPX4 inhibitor and a ferroptosis activator,18,19 was used to determine whether APAP could repair neuronal damage caused by LPS-induced ferroptosis. In this part, HT22 cells were randomly divided into 8 groups according to the following methods: control group, control + APAP group, lipopolysaccharide (LPS) group, LPS + APAP group, control + RSL3 group, APAP + RSL3 group, LPS + RSL3 group, and LPS + APAP + RSL3 group. In the control, APAP and RSL3 groups, HT22 cells were cultivated in DMEM (FBS-free) for 24 h with normal saline, with 50 μM APAP or with 10 μM RSL3. In the APAP + RSL3 group, cells were treated with 50 μM APAP and 10 μM RSL3 together. In the LPS, LPS + APAP, LPS + RSL3, LPS + APAP + RSL3 groups, 1 μg/ml LPS (Cat#L4319, Sigma, USA) was administered, and the cells were cultivated in FBS-free DMEM with 10 μM RSL3, 50 μM APAP, or 10 μM RSL3+50 μM APAP for 24 h after drug concentration screening (CCK-8 method). After 24 h of treatment, cell samples from different groups were collected and used for cell viability detection using the CCK-8 method (n = 3 per group), for GPX4, xCT, 4-HNE protein measurement using the western blot test (n = 6 per group), for ROS determination using the immunofluorescence test (n = 6 per group), for iron content (n = 6 per group), and for TNF-α, IL-6, and HMGB1 measurement using commercial kits (n = 6 per group) (Figure 1(b)).
APAP treatment in vivo
Mice in the APAP and CLP+APAP groups received intraperitoneal injections of 100 mg/kg APAP dissolved in 200 μl sterile saline 1 h before the surgery. They were then administered APAP 100 mg/kg/day continuously until the completion of the test. Meanwhile, the sham and CLP groups received 200 μl (an equivalent volume) saline solution per day until the end of the test. The APAP dose was chosen based on early research demonstrating neuroprotective effects.15,17
Preparation of APAP in vitro
A 40 μM APAP stock solution (Cat# B3532, APExBIO, USA) was prepared as follows: 10 mg APAP was dissolved in 1654 μL DMSO using ultrasound. The stock solution was then diluted to the required concentration before being utilized.
Preparation of RSL3 in vitro
A 1 m
Survival rate observation
As previously described, survival rates (n = 20 per group) were recorded for 7 consecutive days after surgery.
Morris water maze test
The MWM tests were carried out in the same way as in our previous work.20,21 In brief, survivors (n = 10 per group) in each group were subjected to MWM tests 4–10 days after the sham or CLP operation to allow the abdominal wound to heal. The experimental device, which was obtained from XinRuan Information Technology (Shanghai, China), was primarily comprised of a pool, a camera, and the ANY-maze video analysis system. The pool’s inner wall was black, its diameter was 100 cm, and its height was 50 cm. The pool was split into four quadrants, and a 6 cm diameter escape platform was installed in the center of one of the quadrants. Water containing odorless black dye was added to a level 1 cm above the platform, and the temperature of the water was adjusted to 21 ± 2°C. A hidden platform experiment and a spatial exploration exercise were part of the MWM test. The hidden platform experiment began on the 4th day following the sham or CLP procedure and lasted 7 days. The mice were placed in the water, facing the pool wall. The ANY-maze video system captured the escape latency of any mouse that discovered the platform. If the mouse did not reach the platform within 90 s, the escape latency was recorded as 90 s. The platform was then removed on the last day (the 10th), and the spatial exploration experiment was terminated. The mice were placed in the water in the quadrant opposite the platform and permitted to swim freely for 90 s. The number of crossings across the former platform location was counted. Each animal was dried in a container under a heat lamp for 5 min before being placed in its home cage.
H&E staining
Each experimental group’s mice (n = 3) received an intraperitoneal injection of 2% chloral hydrate (400 mg/kg) 24 h after sham or CLP surgery. Mice were transcardially perfused with PBS followed by 4% paraformaldehyde in 0.1
Ultrastructural assay
The hippocampal tissue in different groups (n = 3 per group) was cut into 1 mm × 1 mm × 1 mm sections and fixed in 2.5% glutaraldehyde within 1 min for 24 h. The tissue was then fixed in 1% osmium acid for 1.5 h and routinely dehydrated and embedded, and ultrathin sections were made, stained with uranyl acetate and lead citrate, and scanned with an HT7800 transmission electron microscope (Hitachi, Inc. Ltd, Japan) to observe the mitochondria of hippocampal tissue.
Iron assay (colorimetric) in vivo
According to the kit instructions, hippocampal tissues (n = 6 per group) were taken 24 h after sham or CLP surgery, homogenized, and assayed for hippocampal iron content (Cat#83366, Abcam, Britain).
Iron (ferrozine) in vitro
The cells were washed twice with PBS (resuspension), and 400 μL of lysis solution was added to the cell precipitate, lysed at 4°C for 1 h, and then centrifuged at 4°C for 10 min at 2000 g. The supernatant was transferred to a 96-well plate at 50 μL per well, with 6 wells per sample. Add 50 μL/well of 0. Hydrochloric acid (1 mol/L) was mixed well and cultivated for 30 min at 25°C. Then, 100 μL of the iron probe was added to each well, mixed well, and cultivated for 60 min at 25°C protected from light. Lysis solution: 20 mmol/L Tris-HCl pH 7.5; 150 mmol/L NaCl; 2% (v/v) Triton X-100; 1 mmol/L EDTA. Iron probe: 525 mg ferrozine (Cat# 82,950, Sigma, USA) and 15 g thiourea dissolved in 1 L of 4 mol/L sodium acetate solution.
ROS assay in vivo
Mice (n = 6 per group) were randomly selected at 24 h after modeling, and hippocampal tissues were extracted and processed into single-cell suspensions using enzymatic digestion for cell counting. The fluorescent probe was added strictly based on the instruction manual of the kit (Cat# 139,476, Abcam, Britain), incubated at 37°C for 60 min, centrifuged at 1000×g for 10 min, and washed, and the cell precipitate was collected for fluorescence detection (excitation wavelength: 500; emission wavelength: 525). The quantitative analysis of ROS was evaluated by its ratio to the density of the control group, which was defined as 100%.
ROS assays in vitro
For in vitro ROS analysis, Logarithmic HT22 cells were digested with Trypsin, collected and diluted, and inoculated into 24-well plates with 7000 cells per well. After overnight incubation in an incubator at 37°C with 5% CO2, the cells were confirmed to be in good condition by microscope observation. According to the cells, the density reached 60–70%; the cells were given drugs in each group during the experiment. After 24 h of treatment, a μ5 mol/L dihydroethidium (Cat# R053066, LuoEn, China) or 1 μmol/LBODIPY C11 (CAT# D3861, THERMO, USA) solution was added, and the cells were incubated at 37°C for 30 min for the ROS assay. Five fields were randomly selected and photographed under a fluorescence microscope, and the intensity of red fluorescent cells was recorded. The quantitative analysis of ROS was evaluated by its ratio to the fluorescence intensity of the control group, which was defined as 100%.
Cell viability assay
The vitality of the cells was determined using Cell Counting Kit8 (CCK8) assays (Dojindo Molecular Technologies, Inc. Kumamoto, Japan). The experiment was performed based on the instructions. The cell density was determined using a microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA), and the absorbance was measured at 450 nm. Cell viability was evaluated as a percentage of cytoprotection relative to the control group, which was set to 100%.
Determination of inflammatory cytokines
TNF-α, IL-6, and HMGB1 in the hippocampus (n = 6 per group) and HT22 cells were tested by using an ELISA kit (TNF-α: Cat#RAB0479, Sigma–Aldrich, USA; IL-6: Cat#259341, Abcam, Britain; HMGB1: Cat#BOS-14,703, BOSK, Wuhan, China). The experiment was performed based on the instructions. In brief, 100 μL of the standard or sample was put to each well and incubated at 37°C for 90 min. The particular antibody solution was then added to each well and incubated for 60 min at 37°C.Each well received 100 μL of enzyme conjugate and was incubated at 37°C for 30 min. Following 5 washing, 90 μL of substrate solution was added. After 15 min, 50 μL of terminating solution was added to stop the reaction. Finally, using a fluorescence reader, the optical density of each well was measured at 450 nm and adjusted at 570 nm.
Western blotting
Western blotting was applied to detect the expression of GPX4, xCT, and 4-HNE in the mouse hippocampus (n = 6 per group) and HT22 cells. The proteins were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), transferred to membranes, blocked with skimmed milk, incubated with GPX4 primary antibody (Cat# 125,066, Abcam, Britain), xCT primary antibody (Cat# 175,186, Abcam, Britain), 4-HNE primary antibody (Cat# 46,545, Abcam, Britain) and β-actin primary antibody (1:2000, Cat# A5541, Sigma, USA), and refrigerated overnight at 4°C. The following day, the membranes were washed 5 times with Tris-buffered saline Tween (TBST) for 5 min each, and the goat anti-mouse antibody (1:4000, Cat#ab6789, Abcam, Britain) or goat anti-rabbit antibody (1:4000, Cat#ab6721, Abcam, Britain) was incubated at room temperature for 1 h. The membranes were washed 5 times with TBST for 5 min each time. Enhanced chemiluminescence (ECL) (Cat#WBKLS0100, Merck Millipore, USA). Was added at low light and exposed to the Bio–Rad image analysis system. The gray values of the bands were analyzed by ImageJ software, and the ratio of the gray value of the target protein bands to that of the β-actin bands was used to reflect the expression level of the target protein.
Statistical analysis
Survival rates were reported as a percentage (%), escape latency, swimming speed, and time spent in the target quadrant in MWM tests were displayed as the mean ± standard deviation (SD), and platform crossing times were shown as the median (interquartile range). Others are represented as the mean ± standard deviation (SD). The Long-rank (Mantel–Cox) test with Bonferroni correction was used to examine survival rates. Two-way ANOVA with repeated measurements was used to investigate the interaction between time and group (escape delay and swimming speed in MWM). The post hoc Bonferroni test was performed to examine the differences in escape latency and swimming speed in the MWM. Mann–Whitney U tests were used to investigate platform crossing times. There were no data lost for the MWM and survival rate analyses. Furthermore, if the value in other biochemical data matched the normal distribution, the unpaired t test was used. If the value had a nonnormal distribution, the Mann–Whitney test was used. p < 0.05 was defined as statistically significant. SPSS statistical software (version 22.0) and GraphPad Prism software (version 8.0) were used for statistical analysis.
Results
APAP reduced sepsis-related mortality and cognitive impairment in CLP mice
By observing 7-days survival rates and performing MWM tests on mice with sham or CLP operations, we were able to assess the therapeutic efficacy of APAP. Compared to the sham group, the CLP group’s 7-days postoperative survival rates were considerably lower (p < 0.05 vs. the sham group). When compared to the CLP group, APAP treatment significantly boosted survival rates in the CLP+APAP group (p < 0.05 vs. CLP group) (Figure 2(a)). APAP reduced sepsis-related mortality and cognitive impairment in CLP mice. Mice were given an intraperitoneal injection of 100 mg/kg APAP or an equal volume of normal saline 1 h before the sham or CLP operation, respectively, until the end of the test. (a) Survival rates are depicted as survival percentages (n = 20 per group). (b) Platform crossing time, (c) time spent in the target quadrant and (d–f) escape latency were detected in each group (n = 10 per group). *p < 0.05 vs. sham group; # p < 0.05 vs. CLP group.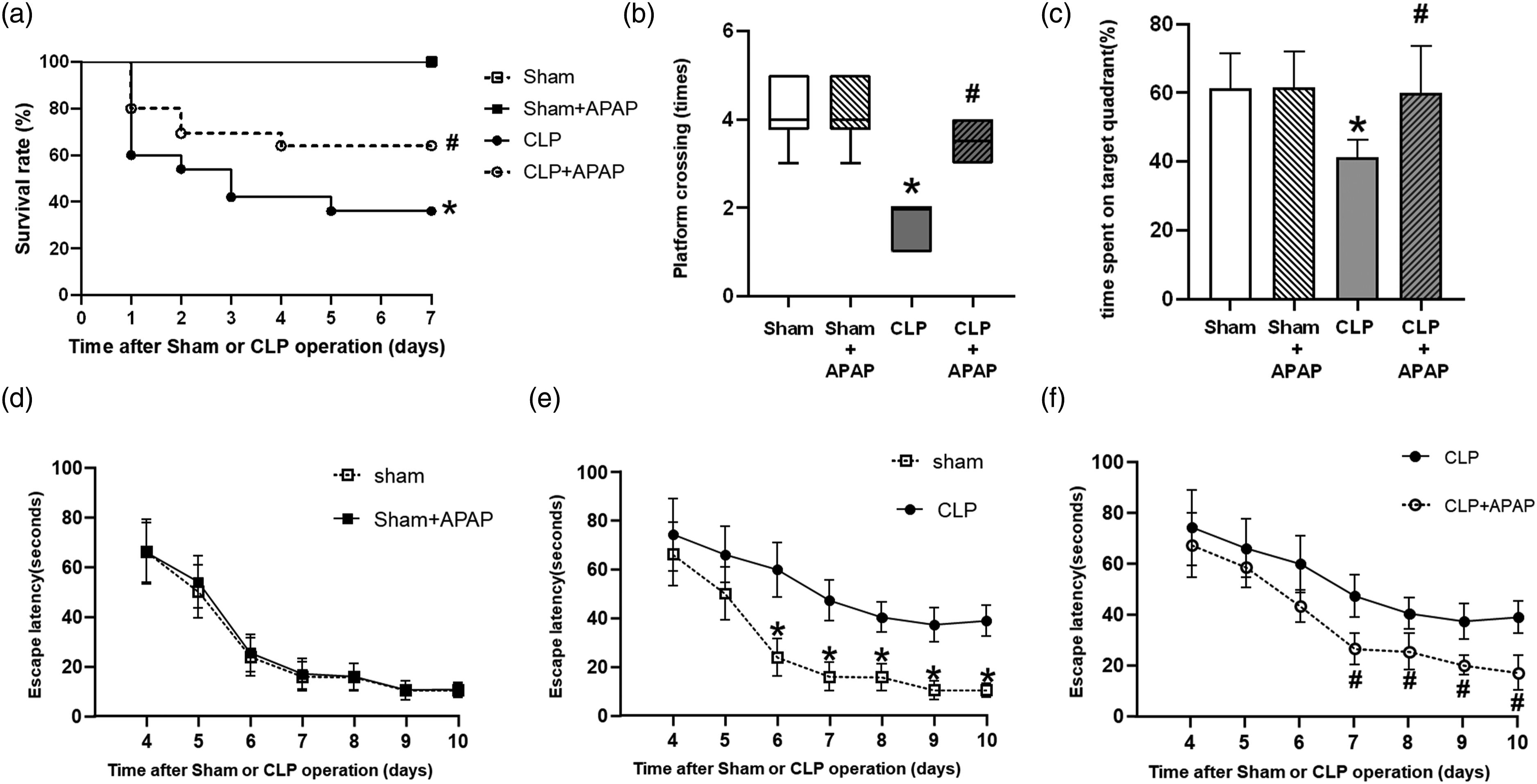
The MWM results showed that the number of platform crossings and the time spent in the target platform quadrant were significantly reduced compared to those in the sham group (p < 0.05 vs. sham group). APAP added the number of times they crossed the platform and the time spent in the target platform quadrant in the CLP + APAP group compared to the CLP group (p < 0.05 vs. CLP group) (Figure 2(b) and (c)). The escape latency in the CLP group was longer than that in the sham group (p < 0.05 vs. sham group). APAP reduced escape latency in the CLP + APAP group compared to the CLP group (p < 0.05 vs. CLP group). There was no difference between the Sham + APAP group and the sham group (p > 0.05 vs. sham group) (Figure 2(d)–(f)). There was no significant difference in swimming speed among groups (p > 0.05).
APAP inhibited ferroptosis in the hippocampal tissue of septic mice via the GPX4 pathway
Previous research has linked ferroptosis to changes in iron concentration and ROS, GPX4, xCT, and 4-HNE levels in tissue.
22
As a result, we compared these markers within each group. First, we used commercial kits to determine the iron concentration and ROS levels. When compared to the Sham group, the iron content and ROS in the hippocampal tissues of mice in the CLP group were significantly higher (p < 0.05 vs. Sham group); when compared to the CLP group, the iron content and ROS in the hippocampal tissues of mice in the CLP+APAP group were lower (p < 0.05 vs. CLP group) (Figure 3(a) and (b)). Then, using western blotting, we determined the expression of GPX4, xCT, and 4-HNE. In comparison to the Sham group, GPX4 and xCT expression in hippocampal tissue was downregulated in the CLP and CLP+APAP groups (p < 0.05 vs. Sham group), 4-HNE expression was upregulated in the CLP group (p < 0.05 vs. Sham group), and there was no statistically significant difference in GPX4, xCT, and 4-HNE expression between the Sham+APAP group and the Sham group (p > 0.05 vs. Sham group) (Figure 3(c)–(f)). APAP inhibited ferroptosis in the hippocampal tissue of septic mice by regulating the GPX4 pathway. The (a) iron content and (b) ROS levels in the hippocampus were detected in each group (n = 6 per group). (c) The expression levels of GPX4, xCT and 4-HNE in the hippocampus were detected by western blot. Quantitative analysis of (d) GPX4, (e) xCT, and (f) 4-HNE is shown as the ratio of band density to that of β-actin. *p < 0.05 vs. sham group; # p < 0.05 vs. CLP group.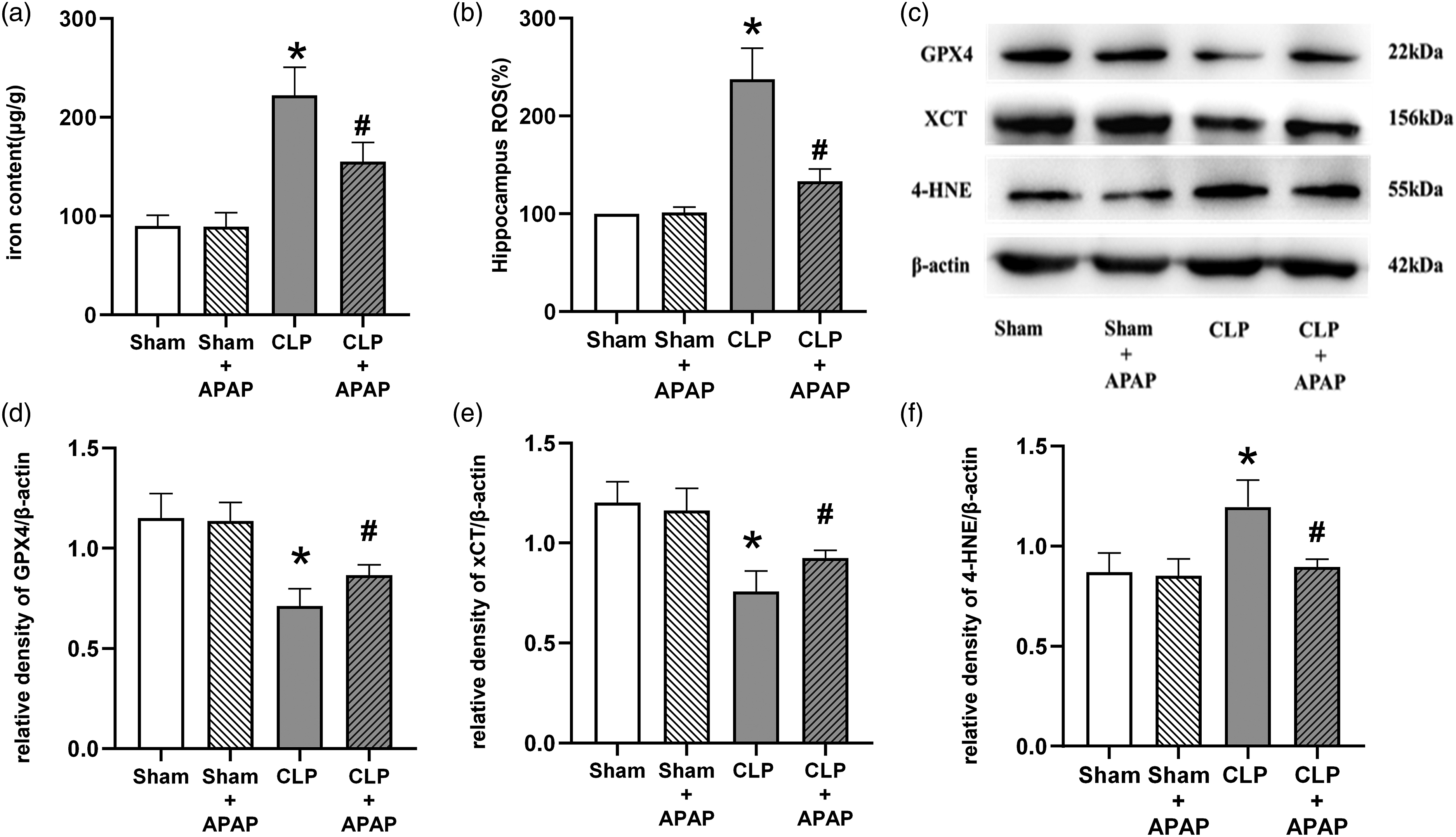
APAP alleviated sepsis-induced hippocampal injury, mitochondrial dysfunction, and neuroinflammation in mice
The hippocampal pyramidal neurons in the CA1 region in the sham and sham + APAP groups were closely arranged with clear structures; the hippocampal pyramidal neurons in the CLP group were disordered, with darkly stained cytoplasm, solidly shrunken nuclei, and unclear structures, while the neurons in the CLP+APAP group were still regularly arranged, with most neurons having normal morphology and a small amount of neuronal damage (Figure 4(a)). APAP alleviated sepsis-induced hippocampal injury, mitochondrial dysfunction, and neuroinflammation in mice. (a) Brain tissue sections were made and stained with hematoxylin and eosin. Arrows point to dead hippocampal neurons. The hippocampal structure was observed (scale bar = 50 μm). (b) The mitochondria of hippocampal tissue were observed by transmission electron microscopy (TEM) (scale bar = 1.0 μm). The proinflammatory cytokines (c) TNF-α, (d) IL-6, and (f) HMGB1 in the hippocampus were detected in each group (n = 6 per group). *p < 0.05 vs. sham group; # p < 0.05 vs. CLP group.
Mitochondrial solidification, increased membrane density, and decreased or missing mitochondrial cristae were observed during ferroptosis. Under electron microscopy, the mitochondrial envelopes of the sham and sham + APAP groups were intact, the mitochondrial crest was evident, and the shape and size were standard. The mitochondrial membrane density rose in the CLP group, the mitochondrial ridge diminished or vanished, and the architecture and volume of the mitochondria were chaotic. Compared to the CLP group, the CLP+APAP group showed reduced mitochondrial membrane density, a distinct mitochondrial crest, typical mitochondrial architecture, and a somewhat more significant volume (Figure 4(b)).
Compared to the Sham group, the levels of TNF-α, IL-6, and HMGB1 in the hippocampal tissues of mice of the CLP group were significantly increased (p < 0.05 vs. Sham group); compared to the CLP group, the TNF-α, IL-6 and HMGB1 levels in the hippocampal tissues of CLP+APAP group were decreased (p < 0.05 vs. CLP group) (Figures 4(c)–(e)).
APAP prevented LPS-induced ferroptosis in HT22 cells by regulating the GPX4 pathway
To investigate whether APAP might rescue neuronal damage from LPS-induced ferroptosis, RSL3, an activator of ferroptosis, was employed. To detect the optimal APAP and RSL3 concentration to protect HT22 cells caused by LPS, the cells were cotreated for 24 h with 0, 25, 50, 100, or 200 μ APAP prevented LPS-induced ferroptosis in HT22 cells by regulating the GPX4 pathway. (a) HT22 cells were incubated with 1 μg/ml LPS and APAP (0–200 μM) simultaneously for 24 h. (b) HT22 cells were incubated with RSL3 (0–10 μM) for 24 h. (c) Cell viability in each group was quantitated by CCK-8 assay (n = 3 per group). The number of control cells that were not exposed to any treatment was defined as 100%. Quantitative analysis of the (d)iron level in HT22 cells is shown as the ratio of the density to that of the control group (n = 6 per group). The ratio of control cells that were not exposed to any treatment was defined as 100%. (e) The expression levels of GPX4, xCT and 4-HNE in HT22 cells were detected by western blot. Quantitative analysis of (f) GPX4, (G) xCT, and (H) 4-HNE is shown as the ratio of band density to that of β-actin (n = 6). *p < 0.05 vs. the control group; #p < 0.05 vs. the LPS group; $p < 0.05 vs. the control+RSL3 group; &p < 0.05 vs. the LPS+APAP group.
Cell viability and iron content were detected 24 h after treatment in different groups. Compared to the control group, LPS incubation substantially increased the iron content but inhibited cell viability in the LPS group (p < 0.05 vs. control group). When compared to the LPS group, APAP reduced the iron content but induced cell viability in the LPS+APAP group (p < 0.05 vs. LPS group). When compared to the RSL3 group, APAP reduced the iron content but induced cell viability in the RSL3+APAP group (p < 0.05 vs. RSL3 group). When compared to the LPS group, RSL3 reduced the iron content but induced cell viability in the LPS+RSL3 group (p < 0.05 vs. LPS group). When compared to the LPS + APAP group, the iron content increased, but cell viability decreased in the LPS + APAP + RSL3 group (p < 0.05 vs. LPS + APAP group) (Figure 5(c) and (d)).
Western blot results showed that GPX4 and xCT were reduced, while the expression of 4-HNE was increased in the LPS group compared with the control group (p < 0.05 vs. control group). When compared to the LPS group, APAP administration increased the levels of GPX4 and xCT while decreasing the level of 4-HNE (p < 0.05 vs. LPS group). When compared to the RSL3 group, APAP administration increased the levels of GPX4 but not xCT while decreasing the level of 4-HNE (p < 0.05 vs. RSL3 group). When compared to the LPS group, RSL3 administration increased the levels of GPX4 but not xCT while decreasing the level of 4-HNE (p < 0.05 vs. LPS group). RSL3 cultivation activated APAP’s protective function. When compared to the LPS + APAP group, the expression of GPX4 was reduced, while the expression of 4-HNE was increased (p < 0.05 vs. LPS + APAP group). There was no difference in xCT expression between the LPS + APAP + RSL3 and LPS + APAP groups (p > 0.05 vs. LPS + APAP group) (Figure 5(e)–(h)).
APAP attenuated oxidative stress and neuroinflammation related to the GPX4 pathway
ROS and proinflammatory factors (TNF-α, IL-6, HMGB1) were increased in the LPS group compared to the control group (p < .05 vs. control group). APAP therapy reduced ROS, TNF-α, IL-6, and HMGB1 levels in the LPS+APAP group compared to the LPS group (p < .05 vs. LPS group). APAP therapy reduced ROS, TNF-α, IL-6, and HMGB1 levels in the RSL3+APAP group compared to the RSL3 group (p < .05 vs. RSL3 group). RSL3 did not reduce ROS, TNF-α, IL-6, or HMGB1 levels in the LPS+APAP group compared to the LPS group (p < .05 vs. LPS group). RSL3 culture revealed APAP’s protective involvement in oxidative stress and neuroinflammation. ROS, TNF-α, IL-6, and HMGB1 levels increased in the LPS+APAP+RSL3 group compared to the LPS+APAP group (p < .05 vs. LPS+APAP group) (Figure 6). APAP attenuated oxidation stress and neuroinflammation related to the GPX4 pathway. Immunofluorescence staining of (a) ROS in HT22 cells (scale bar = 100 μm). Quantitative analysis of (b) ROS in HT22 cells is shown as the relative density of the control group (n = 3 per group). The ratio of control cells that were not exposed to any treatment was defined as 100%. The proinflammatory cytokines (c) TNF-α, (d) IL-6, and (f) HMGB1 in HT22 cells were detected in each group (n = 6 per group). *p < 0.05 vs. the control group; #p < 0.05 vs. the LPS group; $p < 0.05 vs. the control+RSL3 group; &p < 0.05 vs. the LPS+APAP group.
Discussion
SAE is a central nervous system symptom entails a systemic inflammatory response developing to MODS. Patients typically exhibit indicators of brain dysfunction without unequivocal evidence of intracranial infection. 23 The overactivation of inflammatory oxidative stress, along with a dysfunctional blood–brain barrier, is a major cause of cognitive impairment in SAE. 20 In addition, hippocampal neuronal abnormalities have been associated with cognitive dysfunction. 24 Hippocampal neuronal ferroptosis may result in cognitive impairment in sepsis.23,25 Using in vivo and in vitro investigations, we observed that small dose of APAP reduced hippocampal neuronal ferroptosis via the GPX4 pathway for the first time. In animal studies, we discovered that APAP reduced SAE by enhancing 7-days survival cognitive function, lowering hippocampal neuronal ferroptosis, and retraining the inflammatory response. The cell model showed that the protective mechanism of APAP was linked to GPX4 pathway activation.
The pharmacological effects of APAP vary greatly depending on dosage. Excess APAP in the brain causes a substantial drop in glutathione levels, ascorbic acid levels, and SOD activity, resulting in brain damage and serious liver damage. 26 Small dosages of APAP exhibit antioxidative actions in the brain and without liver damage, but larger doses do not.14,15 It has been pointed out that APAP, as an old classic drug for the treatment of sepsis, should continue to explore the relationship between its use and the specific conditions of patients and disease-related factors, and more in-depth exploration of its use and application mechanism is of great significance for the future treatment of sepsis. 27 Our current study indicated that a small amount of APAP decreased the hippocampal expression of iron content, 4-HNE, ROS, TNF-α, IL-6, and HMGB1 but increased the expression of GPX4 and xCT when compared to those of septic mice.
Our studies have observed that small doses of APAP attenuate iron deposition in animal and cellular models. In sepsis, the organism limits pathogen growth by reducing iron uptake by the pathogen. Early on, large amounts of iron can be transferred intracellularly via transferrin, and many inflammatory factors such as IL-6 and TNF-α can increase intracellular storage of iron and reduce the export of intracellular iron outward. This may limit pathogen growth in the short term, but eventually leads to intracellular iron overload, resulting in an iron-mediated Fenton reaction, resulting in cellular lipid peroxidation and cellular ferroptosis. As observed in the study, iron overload was present in both SAE animals and cellular models. In contrast, APAP reduced its mediated lipid peroxidation by reducing iron deposition, reducing cellular and tissue damage.
The primary function of ferroptosis in the formation of particular phospholipid hydroperoxides in the presence of catalytically active iron, which is inhibited by the endogenous systemic xCT/GSH/GPX4 axis; consequently, a disruption in any of these protective compartments might result in ferroptosis. The role of GPX4 as a major regulator of ferroptosis is based on its unique ferroptosis function, which is the reduction of complex hydroperoxides, such as phospholipid hydroperoxides and cholesterol hydroperoxides, to their corresponding counterparts, thereby interrupting the chain reaction of lipid peroxidation. 28 GPX4 is also a critical negative regulator and a downstream component of the ferroptosis pathway in sepsis. According to previous research, it is required to manage bacterial infection and battle ferroptosis in sepsis. 13 Indeed, early studies of conditional deletion of GPX4 in the brain and fibroblasts provided preliminary evidence that neurodegeneration and cell death in hippocampal neurons occur in a nonapoptotic manner requiring significant lipid peroxidation. 29 Inhibition of the GPX4 pathway results in a vast buildup of lipid peroxide and, as a result, a tremendous generation of ROS, resulting in ferroptosis. 9 xCT may transfer glutamate from the cell to the extracellular space and cystine, which may be used to produce GSH, the primary substrate of GPX4. 30 4-HNE, the major ROS metabolite, is a reliable indicator of the degree of lipid peroxidation. 31 Our results showed that APAP increased the expression of ferroptosis core proteins GPX4 and xCT, and decreased the expression of lipid peroxidation indicators 4-HNE and ROS. According to these findings, APAP reduced hippocampus neuronal ferroptosis.
The inflammatory factor IL-6 has anti-infective effects while exacerbating the inflammatory response and can be rapidly elevated in the early stages of inflammation. TNF-α is an important pro-inflammatory mediator in sepsis. HMGB1 is an important inflammatory mediator in the late stages of sepsis. These three inflammatory factors are important for assessing the severity and prognosis of sepsis. The findings showed that APAP attenuated inflammatory factor expression, suggesting that small doses of APAP not only reduced ferroptosis but also reduced inflammation at the same time.
Furthermore, we examined how APAP reduces hippocampal neuronal ferroptosis via the GPX4 pathway. In this section, RSL3, a GPX4 inhibitor that works as a ferroptosis activator, was employed. Because RSL3 includes an electrophilic chloroacetamide, it seems to interact with GPX4’s nucleophilic active site Sec, resulting in irreversible enzyme deactivation. 32 As a result, RSL3 may effectively block GPX4 and indicate that acetaminophen operates via the GPX4 pathway. In the current study, RSL3 reduced the survival of HT22 cells protected by APAP treatment, increased intracellular iron content, 4-HNE, and ROS levels, and decreased GPX4 levels. This finding indicates that APAP protects HT22 cells against ferroptosis via the GPX4 pathway. Unlike erastin, RSL3 inhibits GPX4 directly without affecting its upstream molecular levels; hence, xCT remained unaffected. RSL3 reduced the protective effect of APAP on GPX4,4-HNE and iron levels but not on xCT, which illustrates that the target spot of APAP in the SAE model is GPX4 but not xCT. It is worth noting that following RSL3 injection, the levels of TNF-α, IL-6, and HMGB1 also increased. When the ferroptosis-lowering effect of APAP was reversed, inflammation and oxidative stress were elevated as well.
Inflammation, oxidative stress, and ferroptosis are all intertwined. First, in ferroptosis, iron interacts with (lipoxygenases) LOXs and (cyclooxygenases) COXs in the Fenton reaction, exacerbating lipid oxidation, and its products prostaglandins and leukotrienes enhance sepsis microvascular abnormalities, producing greater inflammatory and oxidative stress. 4 Second, excess iron damages mitochondria, causing inflammatory oxidative stress to worsen. 4 Third, despite attenuating pathogen-associated molecular patterns (PAMPs), attempting to separate pathogenic bacteria and iron results in internal iron overload, resulting in cellular ferroptosis, cell membrane rupture, and the release of substantial quantities of inflammatory factors. 33 In turn, the released inflammatory factors increase ferroptosis, resulting in a vicious cycle. In this investigation, we discovered that APAP might reduce inflammation and oxidative stress by lowering ferroptosis, which may disrupt the vicious cycle of ferroptosis and inflammation. Previously, it has been demonstrated that lowering microglial activity can diminish ferroptosis, which is consistent with our findings. 34 Our study investigates ferroptosis in animal and cellular models of SAE with low doses of APAP for the first time, and found that APAP attenuated ferroptosis and inflammation in hippocampal neurons and reduced cognitive dysfunction in SAE mice. In the cellular model, APAP was explored to exert an inhibitory effect on ferroptosis through the GPX4 pathway and consequently attenuate the inflammatory response. The link between APAP and ferroptosis, inflammation, and oxidative stress in the face of sepsis requires additional exploration.
Conclusions
Finally, we discovered that APAP reduces cognitive impairment in septic mice by inhibiting hippocampal neuronal ferroptosis. The GPX4 signaling pathway was involved in the therapeutic effect. Our findings show that APAP may be a viable treatment for SAE.
Footnotes
Authors’ contributions
Jing Chu: Methodology, Software, Data curation, Writing -original draft. Yi Jiang: Methodology, Data curation. Wenyu Zhou: Software, Validation. Jialei Zhang: Methodology, Software. Hong Li: Data curation, Software. Yang Yu: Conceptualization, Writing - review and editing. Yonghao Yu: Supervision.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Natural Science Foundation of China (grant nos. 82,001,149 and 82,072,150), the Tianjin Health Science and Technology Projects (grant no. KJ20023), the Science and Technology Development Fund of Tianjin Education Commission for Higher Education (grant no. 2019KJ201), and the Tianjin Natural Science Foundation (grant no. 20JCQNJC01050).